
3.3几种典型晶体的结合
分散的原子相互结合成晶体的根本原因在于这些原子结合起来后整个系统具有更低的能量。在结合过程中,有一定的能量W 释放出来,称为晶体的结合能。如果以分散的原子作为计量相互作用势能的零点,则 -W 就是结合成晶体后系统的相互作用势能。
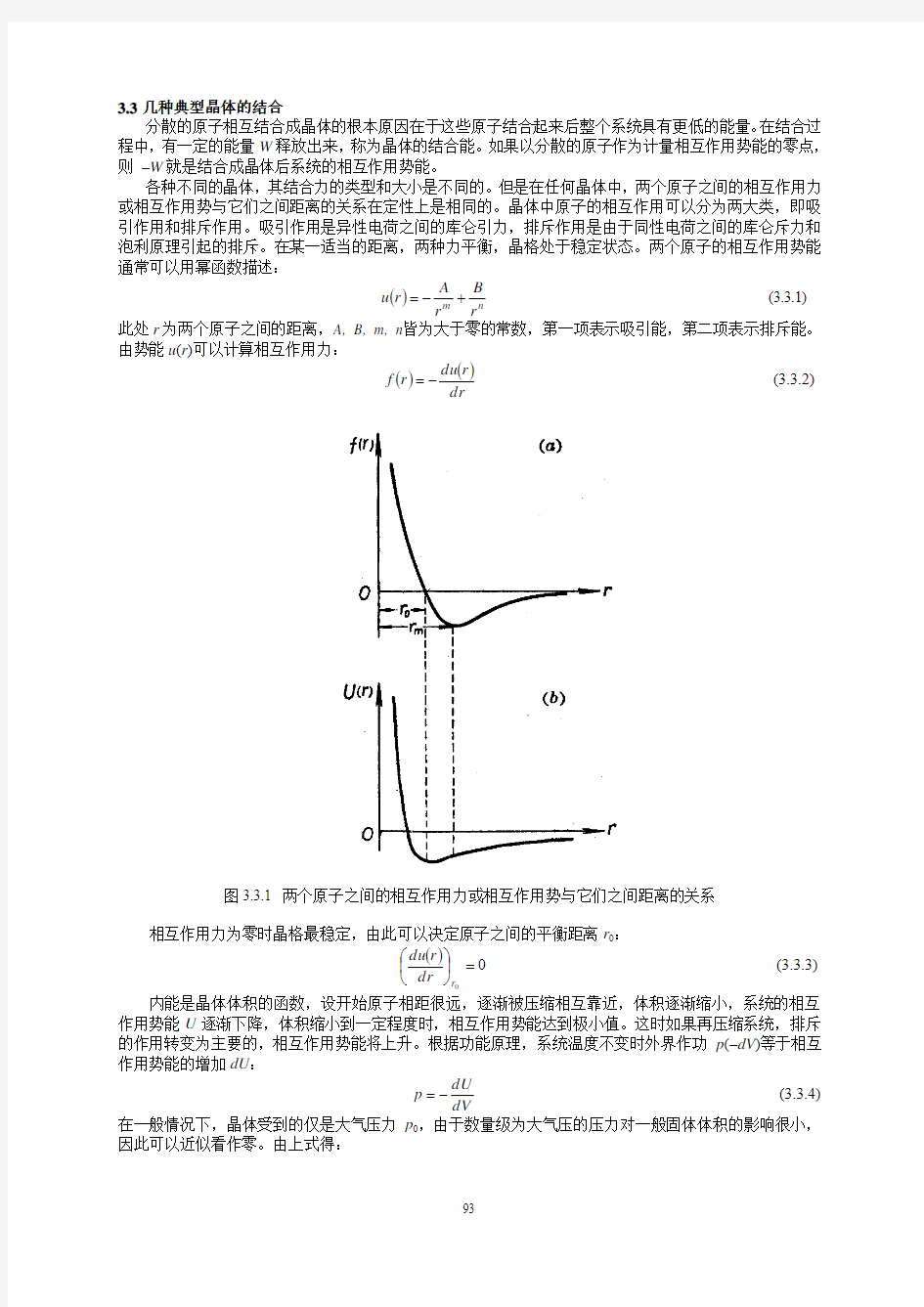
各种不同的晶体,其结合力的类型和大小是不同的。但是在任何晶体中,两个原子之间的相互作用力或相互作用势与它们之间距离的关系在定性上是相同的。晶体中原子的相互作用可以分为两大类,即吸引作用和排斥作用。吸引作用是异性电荷之间的库仑引力,排斥作用是由于同性电荷之间的库仑斥力和泡利原理引起的排斥。在某一适当的距离,两种力平衡,晶格处于稳定状态。两个原子的相互作用势能通常可以用幂函数描述:
()n m r
B
r A r u +-= (3.3.1)
此处r 为两个原子之间的距离,A, B, m, n 皆为大于零的常数,第一项表示吸引能,第二项表示排斥能。由势能u (r )可以计算相互作用力:
()()dr
r du r f -= (3.3.2)
图3.3.1 两个原子之间的相互作用力或相互作用势与它们之间距离的关系
相互作用力为零时晶格最稳定,由此可以决定原子之间的平衡距离r 0:
()00=???
??r dr r du (3.3.3)
内能是晶体体积的函数,设开始原子相距很远,逐渐被压缩相互靠近,体积逐渐缩小,系统的相互
作用势能U 逐渐下降,体积缩小到一定程度时,相互作用势能达到极小值。这时如果再压缩系统,排斥的作用转变为主要的,相互作用势能将上升。根据功能原理,系统温度不变时外界作功p (-dV )等于相互作用势能的增加dU :
dV
dU
p -= (3.3.4)
在一般情况下,晶体受到的仅是大气压力p 0,由于数量级为大气压的压力对一般固体体积的影响很小,因此可以近似看作零。由上式得:
00≈=-p dV
dU
(3.3.5) 这个关系确定了平衡晶体的体积。
体积弹性模量的定义为:
00V dV dp V B ???
??-= (3.3.6)
其中p 为压力,V dV /-为相对体积变化。压缩系数k 是体积弹性模量的倒数。根据pdV dU -=可得:
20V dV U d V B ????
??= (3.3.7) 3 惰性气体晶体
惰性气体原子的电离能很高,因此它们形成最简单的晶体,其中电子分布接近于自由原子的电子分布。原子最外层的电子壳层完全填满,在自由原子中电子电荷的分布是球对称的。晶体中惰性气体原子尽可能紧密地堆积在一起。形成立方密堆积的面心立方结构。只有氦除外。
范德瓦尔斯-伦敦相互作用 在惰性气体晶体中原子间在瞬时相互感生偶极矩,尽管这些感生偶极矩对时间平均为零,但是它们导致惰性气体原子之间微弱的相互吸引。这种相互作用称为范德瓦尔斯-伦敦相互作用。设原子1的瞬时偶极矩为p 1,在r 处产生电场正比于p 1/r 3,在这个电场作用下原子2将感应形成偶极矩p 2为:
312r
p
E p αα== (3.3.8)
其中α为原子的极化率。两个偶极矩之间的相互作用能为:
p p r p r
12
31
26=α (3.3.9) 这时由于相互作用能与p 1的平方成正比,随时间平均不为零。这种相互作用随距离增加下降很快,相互作用很弱。
排斥相互作用 当两个原子相互接近时,它们的电荷分布逐渐交叠,使系统的静电能改变。在间距足够近的情况下,交叠能是排斥性的,这大部分是由泡利不相容原理所引起的。当两个原子的电荷分布交叠时,原属于B 原子的电子倾向于部分占据A 原子的某些态,这些态本来为A 原子所占据;A 原子对B 原子态也有同样的倾向,泡利原理不允许同时占据相同的量子态,因此对于具有闭壳层的原子,其电子分布的交叠只有伴随着部分电子提升到未被占据的高能态才能发生。因此这种电子交叠使系统的总能量增加,而对相互作用能给出排斥性的贡献。
勒纳-琼斯势 通常将间距为r 的两个惰性气体原子间的相互作用势写成:
()???
?
??????? ??-??? ??=6124r r r u σσε (3.3.10)
这就是勒纳-琼斯 (Lennard - Jones) 势。下表给出了N. Bernardes 所推荐的ε和σ值:
如果惰性气体晶体内含有N 个原子,其总的相互作用势能为:
()()???
?
??????? ??-??? ??=661212421r A r A N r U σσε (3.3.11)
由于相互作用势能为两个原子所共有,因此有1/2的因子。r 表示最近邻原子间的距离。A 12、A 6是只与晶体结构有关的求和常数。下表给出几种典型的晶体结构的结果。
对于所有面心立方结构的惰性气体晶体,平衡晶格常数r 0可以从总相互作用势能取极小值的条件得
到:
09.1/0=σr (3.3.12) Ne 、Ar 、Kr 和Xe 的实验观测值分别为1.14、1.11、1.10和1.09,和理论值符合很好,其偏差可以用量子效应来解释。
例题3.3.1 在量子固体中起主导作用的排斥能是原子的零点能。考虑晶态4
e H 的一个粗略的一维模型:每个氦原子局限在一段长为L 的线段上。基态时,这线段取为粒子波函数的半波长。(1) 求每个粒子的零点动能。(2) 推导维持该线段不发生膨胀所需的力。(3) 在平衡时,动能引起的膨胀倾向被范德瓦尔斯相互作用所平衡,粗略地给出最近邻的范德瓦尔斯能为U (L ) = -1.6L -6 ? 10-60 erg ,其中L 以cm 为单位,求L 的平衡值。 解:
(1) 2
2
22222 ,2 ,2mL m k k L k πελπλ ====
(2) 维持该线段不发生膨胀所需的力为:32
2mL
L F k k πε =??-= (3) 范德瓦尔斯相互作用为:760106.9--??=??-=L L
U F vdw
vdw
由动能引起的膨胀倾向被范德瓦尔斯相互作用所平衡vdw k F F =可得:
()37
60
2
2
410
97.7106.9--?=?=
π
m L , L = 0.53 ?
4 离子晶体
离子晶体由正负离子组成,典型的离子晶体如NaCl ,正负离子中正负离子相间排列,使每一种离子以异号的离子为最近邻,因此库仑作用的总效果是吸引的。离子晶体吸引相互作用势能主要就是这种库仑吸引,范德瓦尔斯吸引部分只给出很小的贡献。大约只有百分之一或二。正负离子都具有满的电子壳层结构,当两个满电子壳层的离子相互接近到它们的电子分布发生交叠时,由于泡利原理会产生强烈的排斥作用。
马德隆能 离子晶体相互作用势能的主要贡献是库仑相互作用静电势能,称为马德隆能。以NaCl 晶体为例,正负离子具有球形对称的满壳层结构,考虑库仑作用时可以看作点电荷。先考虑一个处于坐标原点的正离子的平均库仑能。如果令r 表示最近邻离子的距离,这个能量为:
()(
)
∑
++-++3
213
21,,2
/12232
222
21024121
n n n
n n n r
n r
n r n q πε (3.3.13)
很容易验证,凡负离子格点:
=++321n n n 奇数 (3.3.14) 正离子格点:
=++321n n n 偶数 (3.3.15) 计算负离子库仑能的方法是完全相似的,只需要将正离子的结果改变电荷符号,不影响这种能量的结果。因此一对正负离子的能量,或一个原胞中理论值的能量恰好为上述结果的2倍:
()()
r
q n n n
r q n n n
n n
n 02,,2
/123
2221
024143
213
2
1
πεαπε-=++-∑++ (3.3.16)
其中求和为一无量纲数值,完全取决于晶格结构,它是一个负值,因此写成-α,α称为马德隆常数。 例题3.3.2证明两种一价离子组成的一维晶体的马德隆常数为:2 ln 2。 解:
()2ln 24131211212
1
1
=??
?
?????+-+-?=-=∑
∞
+n
n α
中性组合法 马德隆提出了有效地计算此常数的中性组合法。其物理基础为:将离子晶体分成很多完全
相同的电中性区域,以二维NaCl 晶体为例,电中性区域可以取为以某一Cl -离子为中心到任意离子的距离为半边长的正方形区域。半边长为n 倍最短离子间距的中性区称为n 级近似的中性区。选定某一中性区的中心Cl -离子,其它所有中性区域和此中心Cl -离子的相互作用势能可以忽略,只需计算选定中性区域内的所有其它离子和中心Cl -离子之间的相互作用势能,由此确定的马德隆常数随近似级数的增加迅速收敛。一般取2级近似就可以得到足够的精度。
Na +
Cl -
图3.3.2 二维NaCl 晶体
对二维NaCl 晶体,二级近似具体计算如下:
则马德隆常数为:
∑
=
d
zm γ
α=1.607 几种常见的三维离子晶体的马德隆常数为:1.748 (NaCl), 1.763 (CsCl), 1.638 (ZnS)。
相互作用能 设离子晶体包含N 个原胞,综合考虑到马德隆能和交叠排斥能,总相互作用势能为:
()???
???+-=??
????+-=n n r B r A N r B r q N r U 024πεα (3.3.17)
由相互作用能取极小值的条件可得平衡但是最近邻距离r 0:
010
20=-+n r nB
r A (3.3.18)
因此可得A 和B 之间的关系,代入总相互作用势能公式可得离子晶体结合能为: ()??? ?
?
-=
-=n r q N r U W 1140
02
0πεα (3.3.19)
几种典型的离子晶体的排斥势能常数n 的数值为:7.77 (NaCl), 8.09 (BaBr), 8.69 (KCl), 8.85 (KBr), 9.13 (RbCl)和9.00 (RbBr)。由此可见总相互作用势能主要取决于库仑能,排斥能只占库仑能的1/n 。 5 共价晶体
共价结合的特点 共价晶体是由共价键结合起来的晶体。共价结合具有的两个基本特征:饱和性和方向性。饱和性是指一个原子只能形成一定数目的共价键。方向性是指原子只在特定方向上形成共价键。 轨道杂化 原子在形成共价键时,可以发生轨道杂化。碳原子有6个电子,基态的电子组态为1s 22s 22p 2,
在金刚石中每个碳原子与4个近邻碳原子形成共价键结合。它们不是以基态波函数为基础的,而是由2s 和2p 波函数组成的杂化波函数形成共价键:
()z y x p p p s 2222121
????ψ+++=
()z y x p p p s 2222221
????ψ--+=
()z y x p p p s 2222321
????ψ-+-=
()
z y x p p p s 222242
1
????ψ+--= (3.3.20)
这些杂化轨道的特点是它们的电子密度分布分别集中在四面体的4个顶角方向。形成4个共价键,当然,电子处于杂化轨道上,能量比碳原子的基态提高了,但是经过杂化以后,成键的数目增多了,而且由于电子密度更加密集在四面体顶角方向上,使得成键能力更强了,形成共价键时能量的下降足以补偿轨道杂化所增加的能量。
对于立方硫化锌结构,类似于金刚石结构,但是基元中的两个原子不同,所形成的共价键包含有离子键的成份。以重要的半导体材料GaAs 为例,它们的离子实分别为带+3e 的离子Ga 3+和+5e 的离子As 5+。每一对Ga 和As 有8个电子。如果是完全的共价结合,共价键上的每对电子均分在两个邻近的原子上,总起来看,Ga 原子有一个负电荷,As 原子有一个正电荷,形成Ga 1-As 1+。如果是完全的离子结合,意味着Ga 原子有3个价电子转移到As 原子上,形成Ga 3+As 3-。而实际情况是介于二者之间的。通常引入有效离子电荷q *,可以用成键态波函数讨论有效离子电荷。波函数()B A c λ??ψ+=意味着在A 原子和B 原
子上电子的几率P A 和P B 分别为:
2
11
λ+=
A P (3.3.21)
2
2
1λ
λ+=B P (3.3.22) 因此Ga 原子 (B 原子) 和As 原子 (A 原子) 的有效电荷分别为为:
???
? ??+-=*22183λλB
q (3.3.23)
??
? ??
+-=*21185λA q (3.3.24) 原则上,如果能求出λ的值就可以得到有效离子电荷的数值,也可以由实验测定。 电离度和原子的负电性 通常引入电离度 (ionicity) 来描述共价结合中离子性的成份。有3种不同标度电离度的方式。卡尔森 (C. A. Coulson) 定义电离度f i 为:
2
11λλ+-=+-=B A B A i P P P P f (3.3.25)
显然对于完全的共价结合λ = 1,f i = 0;对于完全的离子结合λ = 0,f i = 1。当共价结合中含有离子键成份时,f i 介于0和1之间,f i 数值越大,表明离子键成份越多。
泡令 (L. Pauling) 根据原子的负电性定义了另一种电离度的标度方式:
()4/2
1B A x x i
e f ---= (3.3.26)
其中x A 和x B 分别为原子A 和原子B 的负电性。原子的负电性定义为:
负电性 = 0.18 ? (电离能 + 亲和能) (单位为 eV) (3.3.27) 电离能为原子失去一个电子所必须的能量,亲和能是一个中性原子获得一个电子成为负离子时所放出的能量。系数0.18的选择只是为了使Li 的负电性为1,没有原则上的意义。
菲利蒲 (J. C. Phillips) 把成键态和反键态之间的能隙E g 看成是共价结合成份的贡献E h 和离子结合成份的贡献C 的组合,它们之间的关系为:
22
2C E E h g += (3.3.28) E h 和C 可以从光学系数的实验结果中分析得到。菲利蒲定义电离度为: 2
C E C f h i +=
(3.3.29)
共价结合晶体结合能的理论计算远比离子晶体复杂。一般用Hohenberg-Kohn 和Kohn-Shen 局域密度
泛函理论计算,对各类半导体材料的结合能、晶格常数和体积弹性模量计算的结果,与实验符合得相当好。
离子半径 在量子力学中,电子在空间某一点出现的几率与在该点的电子波函数模的平方成正比。这样很难确定离子的半径。但是,在单质金属中,可以通过X 射线衍射测定相邻两原子间的距离,可以假定金属原子半径为此距离的一半。但是对离子晶体来说,通过X 射线衍射测定相邻两离子间的距离后,还是很难确定正负离子的半径。根据实验结果,NaF 和KF 晶体的离子间距分别为2.31?和2.66 ?,相差0.35 ?,NaCl 和KCl 晶体的离子间距相差0.33 ?,NaBr 和KBr 晶体的离子间距相差0.32 ?。这个差数近似为一常数,可以假定这个差数就是钠离子和钾离子的半径差值。看来离子似乎有一个确定的半径。
常用的离子半径有高希米特(Goldschmidt)半径,泡林(Pauling)半径和察卡里逊(Zachariasen)半径。 泡林将具有相同电子数的离子归为一个等电子系列,例如:N ---, O --, F -, Ne, Na +, Mg ++, Al +++, ? ? ?。他认为离子半径主要取决于最外层电子的分布,而对于等电子离子,离子半径还与作用于其上的有效核电荷(Z - S )出正比:
S
Z C R n
-=1 (3.3.30)
式中R 1称为单价半径,C n 是由外层电子主量子数n 所决定的一个常数,S 为屏蔽常数,由实验确定,Z 为原子序数。
例如,NaF 的离子间距由X 射线衍射测定为2.31 ?,, Na +和F -属Ne 的等电子离子,这种结构的屏蔽常数S = 4.52,于是有:
???
?
?
????
=+=-==-=-+-+ ? 31.248.452.4948.652.411F N n n F n n N R R C C R C C R a a (3.3.31)
解此方程组可得:? 95.0=+a
N R ,? 36.1=-F R ,C n = 6.2。泡林还利用KCl, RbBr, CsI, 按照相同的方法计
算得到了一系列的单价半径,再由公式:
()1/21--?=n R R ηη (3.3.32) 换算成多价离子的晶体半径ηR ,式中n 为离子晶体的晶格参量玻恩指数,η为离子的价数。下表为泡林单价半径、晶体半径和高希米特半径的对照表:(单位为?)
察卡里逊给出了离子间距D N ,正、负离子半径R +、R -的关系:
N N R R D ?++=-+ (3.3.33) 其中N ?是配位数为N 的校正值,室温时为:
N 1 2 3 4 6 8 12
N ? -0.50 -0.31 -0.19 -0.11 0 0.08 0.19
4 金属晶体
原子结合成金属时,原来属于各原子的价电子不再束缚在原子上,而转变为在整个晶体中运动,它们
的波函数遍及整个晶体。金属的导电性、导热性等都是和共有化电子可以在整个晶体中自由运动有关的。金属结合的特征性质是金属中价电子的动能与自由原子相比有所降低。碱金属可以想象为淹没在近均匀负电荷海洋中的正电荷阵列。负电子云和正离子实之间存在库仑吸引作用,显然体积越小,库仑吸引作用就越强,库仑相互作用的能量越低。排斥作用有两个来源:当体积缩小,共有化电子密度n 增加的同时,它们的动能也增加,根据托马斯-费米统计,动能正比于n 2/3;另一方面,当原子实相互接近到它们的电子云发生显著交叠时,由于泡利原理会产生强烈的排斥作用。晶体的平衡是依靠库仑吸引和这两种排斥作用决定的。
由于金属结合没有方向性和饱和性,金属结合首先是一种体积效应,原子愈紧凑,库仑能就愈低。因此很多金属元素采取面心立方或六角密积结构,它们都是排列最紧密的晶体结构,体心立方也是比较普通的金属结构。
金属结合能也可以用泛函理论来计算,对各种金属材料的结合能、晶格常数和体积弹性模量计算的结果,与实验符合得相当好。
习题
3.3.1 由N 个原子组成的晶体的相互作用势能可以表示为:
()??? ??+-=n m r r
N r U βα 求 (1) 平衡间距r 0 (2) 结合能W
(3) 体积弹性模量B
(4) αβ,的值。取m = 2,n = 10, r 0 = 3 ?,W = 4N eV 。 3.3.2 由气相测量结果得到H 2的勒纳德-琼斯参数为ε = 50 ? 10-16 erg ,σ = 2.96 ?。计算面心立方结构H 2
晶体的结合能。氢分子可以作为球形处理。结合能的实验值为0.751 kJ / mol ,将计算值与测量值进行比较。
3.3.3 说明使用中性组合法计算马德隆常数的物理基础。用中性组合法计算三维维NaCl 晶体的马德隆常
数。取二级近似。
3.3.4 经过sp 3杂化后形成的共价键,其方向沿正四面体四个顶角方向,求共价键之间的夹角。
3.3.5 讨论使离子电荷加倍引起的对NaCl 晶格常数和结合能的影响。假定排斥势不变,并取n = 9。
一、 计算方法 密度泛函理论(DFT )、含时密度泛函理论(TDDFT ) 二、 计算方法原理 1. 计算方法出处及原理 本计算方法设计来源于量子化学理论中的Born –Oppenheimer 近似,给近似下认为原子核不动, 这样电子就相当于在一个由核产生的外部的静态势场 V 中运动。那么一个固定的电子态可以用波函数 Ψ(1r , · · · ,N r ), 并且满足多 N 电子体系薛定谔方程: ()() 22????,2N N N i i j i i i i j H T V U V r U r r E m
333*231212()(,,)(,,) N N N n r N d r d r d r r r r r r r =???ψ???ψ?????? (2-4) 更重要的是, DFT 的核心理念告诉我们, 对于一个给定的基态, 如果基态 的电子密度0()n r 是知道的话, 那么基态的波函数012(,,)N r r r ψ???就唯一确定。也就是说, 基态的波函数0ψ是基态电子密度0n 的泛函[11], 表达为: [] 00n ψ=ψ (2-5) 既然有以上的假定, 那么对于基态的任何一个观测量?O , 它的数学期望就应该是0n 的泛函: [][][]000 ?O n n O n =ψψ (2-6) 特别的, 基态的能量也是0n 的泛函: [][][]0000 ???E E n n T V U n ==ψ++ψ (2-7) 这里外部势能的贡献[][]00?n V n ψψ可以通过基态的电子密度0 n 来精确表达: 300[]()()V n V r n r d r =? (2-8) 或者外部势能?V ψψ可以用电子密度 n 来表达: 30[]()()V n V r n r d r =? (2-9)
密度泛函理论及其应用 一、密度泛函理论(Density Functional Theory :DFT ) VASP 的理论基础是电荷密度泛函理论在局域电荷密度近似(LDA )或是广 义梯度近似(GGA )的版本。DFT 所描述的电子气体交互作用被认为是对大部分 的状况都是够精确的,并且它是唯一能实际有效分析周期性系统的理论方法。 1.1 单电子薛定谔方程式 一个稳定态(与时间无关)的单一粒子薛定谔方程式可表示为一个本征值问 题(暂略动能项的 /2m ): ()()H r E r ψψ= (1) 2[]()()V r E r ψψ-?+= (2) 多体量子系统 (如双电子的薛定谔方程式): 2212121212[(,)](,)(,)V r r r r E r r ψψ-?-?+= (3) 在普遍的状况下,12(,)V r r 里的12,r r 是无法分离变量的,因此,即便简单如 双电子的薛定谔方程式就己经没有解析解了。而任何的计算材料的量子力学问 题,都需要处理大量数目的电子。 1.2 Hohenberg-Kohn 定理 量子力学作为20世纪最伟大的发现之一,是整个现代物理学的基石。量子力 学最流行的表述形式是薛定谔的波动力学形式,它的核心是波函数及其运动方程 薛定谔方程。对一个给定的系统,我们可能得到的所有信息都包含在系统的波函 数当中。对一个外势场v (r)中的N 电子体系,量子力学的波动力学范式可以表示 成: v (r) ?Ψ (r1; r2; …; r N ) ?可观测量 (4) 即,对给定的外势,将其代入薛定谔方程可以得到电子波函数,进一步通过
波函数计算力学量算符的期望值可以得到所有可观测量的值。电荷密度是这些可 观测量中的一个: 333* 232()...(,...)N N n r N d r d r d r r r r =ψ???2(,...)N r r r ψ (5) 如前所述,任何的计算材料的量子力学问题,都需要处理大量数目的电子。 而,对于超过两个电子以上的体系,薛定谔方程就已经难以严格求解了。对于实 际物质的这样一种每立方米中有2910数量级的原子核和电子的多粒子系统,我们 是更不可能由薛定谔方程来严格求解其体系的电子结构的。但,建立于 Hohenberg-Kohn 定理上的密度泛函理论不但给出了将多电子问题简化为单电子 问题的理论基础,同时也成为分子和固体的电子结构和总能量计算的有力工具。 因此,密度泛函理论是多粒子系统理论基态研究的重要方法。 密度泛函理论的基本想法是原子、分子和固体的基态物理性质可以用粒子密 度函数来描述,这源于H.Thomas 和E·费米1927年的工作。密度泛函理论基础是建 立在P.Hohenberg 和W.Kohn 的关于非均匀电子气理论基础上的,它可归结为两个 基本定理: 定理一:不计自旋的全同费米子系统的基态能量是粒子数密度函数()n r 的唯 一泛函。 它的推论是,任何一个多电子体系的基态总能量都是电荷密度()n r 的唯一泛 函,()n r 唯一确定了体系的(非简并)基态性质。 由于电荷密度与电子数N 直接联系:()n r dr N =?,这样决定多电子薛定谔 方程解的电子数N 和外势场都由电荷密度()n r 唯一确定,因此基态波函数[] F n 以及其它的电子结构性质都由电荷密度唯一确定。 由于()V r 决定了哈密顿量,多电子体系的基态ψ是()n r 的唯一泛函,自然 动能和库仑能也是()n r 的泛函,那么体系的所有性质也将是基态密度的泛函。于 是定义一个普适泛函[]F n ,有: 2,,22,()1 (1)()2()l ps l ps l l ps d r l l V r E r dr r ??Φ+??=+-????Φ?????? []??()F n r T U ≡<ψ+ψ> (6) 适用于任何外场下的具有任意电子数的体系。所以系统基态的能量可表示为
第四章 密度泛函理论(DFT)
4.1 引言 4.2 DFT的优点 4.3 Hohenberg-Kohn定理 4.4 能量泛函公式 4.5 局域密度近似 4.6 Kohn-Sham方程 4.7 总能Etot表达式 4.8 DFT的意义 4.9 小 结
1
4.1 引言
1。概述 ? DFT = Density Functional Theory (1964): 一种用电子密度分布n( r)作为基本变量,研究多粒子 体系基态性质的新理论。 W. Kohn 荣获1998年Nobel 化学奖 ? 自从20世纪60年代(1964)密度泛函理论(DFT) 建立并在局域密度近似(LDA)下导出著名的Kohn -Sham (沈呂九)(KS)方程以来,DFT一直是凝聚态 物理领域计算电子结构及其特性最有力的工具。
2
2。地位和作用 ? 近几年来,DFT同分子动力学方法相结合, 有许多新发展; ? 在材料设计、合成、模拟计算和评价诸多方 面有明显的进展; ? 已成为计算凝聚态物理、计算材料科学和计 算量子化学的重要基础和核心技术; ? 在工业技术领域的应用开始令人关注。
3
4.2 DFT的优点
? 它提供了第一性原理或从头算的计算框 架。在这个框架下可以发展各式各样的能 带计算方法。 ? 在凝聚态物理中,如: 材料电子结构和几何结构, 固体和液态金属中的相变等。 ? 这些方法都可以发展成为用量子力学方法 计算力的, 精确的分子动力学方法。
4
密度泛函理论的进展与问题 摘要:本文综述了密度泛函理论发展的基础及其最新进展,介绍了求解具体物理化学问题时用到的几种常用的数值计算方法,另外对密度泛函理论的发展进行了展望。密度泛函理论的发展以寻找合适的交换相关近似为主线,从最初的局域密度近似、广义梯度近似到现在的非局域泛函、自相互作用修正,多种泛函形式的相继出现使得密度泛函理论可以提供越来越精确的计算结果。另外,在密度泛函理论体系发展的同时,相应的数值计算方法的发展也非常迅速。随着密度泛函理论本身及其数值方法的发展,它的应用也越来越广泛,一些新的应用领域和研究方向不断涌现。 关键词:密度泛函数值计算发展应用 1 研究背景 量子力学作为20世纪最伟大的发现之一,是整个现代物理学的基石。量子力学最流行的表述形式是薛定谔的波动力学形式,核心是波函数及其运动方程薛定谔方程。对一个外势场v(r)中的N电子体系,量子力学的波动力学范式可以表示成: 即对给定的外势,将其代入薛定谔方程可以得到电子波函数,可以得到所有可观测量的值。 当用量子力学处理真实的物理化学体系时,传统的波动力学方法便显得有点力不从心。因为在大多数情况下,人们只是关心与实验相关的一部分信息,如能量、密度等。所以,人们希望使用一些较简单的物理量来构造新的理论[1]。 电子密度泛函理论是上个世纪60年代在Thomas-Fermi理论的基础上发展起来的量子理论的一种表述方式。传统的量子理论将波函数作为体系的基本物理量,而密度泛函理论则通过粒子密度来描述体系基态的物理性质。因为粒子密度只是空间坐标的函数,这使得密度泛函理论将3N 维波函数问题简化为3维粒子密度问题,十分简单直观。另外,粒子密度通常是可以通过实验直接观测的物理量。粒子密度的这些优良特性,使得密度泛函理论具有诱人的应用前景。 2 密度泛函理论的基础 Thomas-Fermi模型 1927 年Thomas和Fermi分别提出:体系的动能可以通过体系的电子密度表达出来。他们提出了一种的均匀电子气模型,把空间分割成足够小的立方体,通过在这些立方体中求
量子力学泛函计算 纪岚森 (青岛大学物理科学学院材料物理一班) 摘要:文章叙述了密度泛函理论的发展,密度泛函理论以“寻找合适的交换相关为主线,从 最初的局域密度近似,,从最初的局域密度近似、广义梯度近似到现在的非局域泛函、自相 互作用修正,多种泛函形式的出现,是的密度泛函在大分子领域的计算越来越精确。近年来 密度泛函理论在含时理论与相对论方面发展也很迅速。计算体系日臻成熟,而我所参加的创 新实验小组就是以密度泛函研究大分子体系。在量子力学泛函计算的产生,发展,理论,分 支,前景等方面予以介绍,本着科学普及的态度希望大家能够更加进一步的理解泛函计算。 关键字:量子力学泛函计算,发展,理论分支,前景,科普 1引言:随着量子理论的建立和计算机技术的发展,人们希望能够借助计算机对微观体系的量子力学方程进行数值求解【3】,然而量子力学的基本方程———Schirdinger 方程的求解是极其复杂的。克服这种复杂性的一个理论飞跃是电子密度泛函理论(DFT)的确立电子密度泛函理论是上个世纪60 年代在Thomas-Fermi 理论的基础上发展起来的量子理论。与传统的量子理论向悖,密度泛函理论通过离子密度衡量体系的状态,由于离子密度只是空间的函数,这样是就使得解决三维波函数方程转化为解决三维密度问题,使得在数学计算上简单了很多,对于定态Schirdinger 方程,我们只能解决三维氢原子,对于更加复杂的问题,我们便无法进行更为精确的计算,而且近似方法也无法是我们得到更为精确的结果。但是密度泛函却在这方面比较先进,是的大分子计算成为可能。【2】 2.过程:第一性原理,密度泛函是一宗量子力学重头计算的计算方法,热播呢V啊基于密度泛函的理论计算成为第一性原理——first-principles。经过几十年的发展密度泛函理论被广泛的应用于材料,物理,化学和生物等科学中,Kohn也由于其对密度泛函理论的不可磨灭的先驱性贡献获得了诺贝尔化学奖。密度泛函理论体系包括交换相关能量近似,含时密度泛函。 3.密度泛函理论的发展: 1交换相关能,在密度泛函理论中我们把所有近似都归结到交换相关能量一项上,所以密度泛函的精确度也就是由交换相关能一项上。寻求更好的更加合适的相关近似,即用相同密度的均匀电子气交换相关泛函作为非均匀系统的近似值,或许这也出乎人们的意料,这样一个简单的近似却得到了一个极好的结论。直接导致了后来的泛函理论的广泛应用。由此获
密度泛函理论
摘要:介绍了密度泛函理论的发展与完善,运用密度泛函理论研究了钒(Vanadium)在高压下的结构相变。通过计算体心立方结构的钒在不同压强下剪切弹性系数C44,发现当压强约95 GPa时C44<0,说明体心立方结构的钒在此条件下是不稳定的。进一步计算分析得到钒在高压下发生了从体心立方到菱面体的结构相变,相变压强约70 GPa,这一结果与实验结果符合。还首次发现当压强约380 GPa时,将会发生菱面体到体心立方的结构相变,这有待实验的验证。 引言:相变的研究受到广泛重视,通过相变研究可以认识物质的内部结构,可以了解原子核的内部性质。尤其是极端条件下—高温、高压下相变的研究一直是人们关注的热点,能量很高的重离子反应能形成高温、高密的区域,在这种条件下会出现许多奇异现象[1]。原子在高压下也会出现许多新的特征,如发生结构相变。过渡金属钒由于有较高的超导转变温度Tc,最近成为实验和理论研究的主题[2—8]。Ishizuka等[2]对钒的实验研究发现:常压下钒的转变温度Tc为5.3 K,并随压强成线性增长的关系,当压强为120 GPa时Tc=17.2 K(迄今是金属中最大的Tc),但压强大于
120 GPa,Tc出现了反常,即不再随压强成线性增长而保持不变。Takemura等[8]对高压下的钒进行了X射线衍射实验,结果显示状态方程并没有奇异性,体心立方结构的钒在压强达到154 GPa 时仍是稳定的。Suzuki和Ostani利用第一性原理对进行了计算,发现横向声子模在加压下有明显的软化,当压强约130 GPa时变成虚的,能说明可能发生了结构相变,但并未给出相变细节[3]。Nirmal等[4]理论计算表明,压强约140 GPa时会发生体心立方到简立方(sc)的结构相变。Landa 等[5,6]计算了体心立方结构的钒在加压下剪切弹性系数C44的大小,发现压强约200 GPa时会出现力学不稳定,并用费米面嵌套解释了不稳定的原因,但并没有给出相变后的结构。最近Ding 等[7]在常温下首次从实验上得到当准静压约63 GPa时钒会发生从体心立方到菱面体的结构相变,并分析了产生结构相变的原因。他们认为,排除传统的s-d电子跃迁的驱动,相变可能与来自于费米面嵌套、带的Jahn-Teller扭曲以及电子拓扑跃迁等因素有关。 基于如上原因,本文运用密度泛函理论研究钒在高压下的结构相变,即通过计算体心立方结构的
1分子(或原子)间相互作用势简介 分子(或原子)间相互作用势的准确性对计算结果的精度影响极大,但总的来说,原子之间的相互作用势的研究一直发展得很缓慢,从一定程度上制约了分子动力学在实际研究中的应用.原子间势函数概念本身已把电子云对势函数的贡献折合在内了,原子间势函数的发展经历了从对势,多体势的过程.对势认为原子之间的相互作用是两两之间的作用,与其他原子的位置无关,而实际上,在多原子体系中,一个原子的位置不同,将影响空间一定范围内的电子云分布,从而影响其他原子之间的有效相互作用,故多原子体系的势函数更准确地须用多体势表示. 2 力场简介
图1 键伸缩势示意图图2键伸缩势示意图
图3二面角扭曲势示意图 在分子动力学模拟的初期,人们经常采用的是对势.应用对势的首次模拟是Alder和Wainwright在1957年的分子动力学模拟中采用的间断对势.Rahman在1964年应用非间断的对势于氩元素的研究,他和Stillinger在1971年也首次模拟了液体HzO分子,并对分子动力学方法作出了许多重要的贡献,比较常见的对势有以下几种: (a)间断对势 Alder和Wainwrigh在1957年使用间断对势 这个势函数虽然很简单,但模拟结果给人们提供了许多有益的启示.后来他们又采取了另一种形式的间断对势。 (b)连续对势 对势一般表示非键结作用,如范德瓦耳斯作用;常见的表达方式有以下几种:
ij ij 其中,Lennard —Jones 势是为描述惰性气体分子之间相互作用力而建立的,因此它表达的作用力较弱,描述的材料的行为也就比较柔韧.也有人用它来描述铬、钼、钨等体心立方过渡族金属.Born-Lande 势是用来描述离子晶体的. Morse 势与Johnson 势经常用来描述金属固体,前者多用于Cu ,后者多用于 Fe .Morse 势的势阱大于Johnson 势的势阱,因此前者描述的作用力比后者强,并且由于前者的作用力范围比后者长,导致Morse 势固体的延性比Johnson 势固体好.对势虽然简单,得到的结果往往也符合某些宏观的物理规律,但其缺点是必然导致Cauchy 关系,即Cl2=C44,而一般金属并不满足Cauchy 关系,因此对势实际上不能准确地描述晶体的弹性性质
密度泛函理论, Density functional theory (DFT)是一种研究多电子体系电子结构的量子力学方法。密度泛函理论在物理和化学上都有广泛的应用,特别是用来研究分子和凝聚态的性质,是凝聚态物理和计算化学领域最常用的方法之一。 理论概述 电子结构理论的经典方法,特别是Hartree-Fock方法和后Hartree-Fock方法,是基于复杂的多电子波函数的。密度泛函理论的主要目标就是用电子密度取代波函数做为研究的基本量。因为多电子波函数有个变量(为电子数,每个电子包含三个空间变量),而电子密度仅是三个变量的函数,无论在概念上还是实际上都更方便处理。 虽然密度泛函理论的概念起源于Thomas-Fermi模型,但直到Hohenberg-Kohn定理提出之后才有了坚实的理论依据。Hohenberg-Kohn第一定理指出体系的基态能量仅仅是电子密度的泛函。 Hohenberg-Kohn第二定理证明了以基态密度为变量,将体系能量最小化之后就得到了基态能量。 最初的HK理论只适用于没有磁场存在的基态,虽然现在已经被推广了。最初的Hohenberg-Kohn定理仅仅指出了一一对应关系的存在,但是没有提供任何这种精确的对应关系。
正是在这些精确的对应关系中存在着近似(这个理论可以被推广到时间相关领域,从而用来计算激发态的性质[6])。密度泛函理论最普遍的应用是通过Kohn-Sham方法实现的。在Kohn-Sham DFT的框架中,最难处理的多体问题(由于处在一个外部静电势中的电子相互作用而产生的)被简化成了一个没有相互作用的电子在有效势场中运动的问题。这个有效势场包括了外部势场以及电子间库仑相互作用的影响,例如,交换和相关作用。处理交换相关作用是KS DFT中的难点。目前并没有精确求解交换相关能的方法。最简单的近似求解方法为局域密度近似(LDA)。LDA近似使用均匀电子气来计算体系的交换能(均匀电子气的交换能是可以精确求解的),而相关能部分则采用对自由电子气进行拟合的方法来处理。 自1970年以来,密度泛函理论在固体物理学的计算中得到广泛的应用。在多数情况下,与其他解决量子力学多体问题的方法相比,采用局域密度近似的密度泛函理论给出了非常令人满意的结果,同时固态计算相比实验的费用要少。尽管如此,人们普遍认为量子化学计算不能给出足够精确的结果,直到二十世纪九十年代,理论中所采用的近似被重新提炼成更好的交换相关作用模型。密度泛函理论是目前多种领域中电子结构计算的领先方法。尽管密度泛函理论得到了改进,但是用它来恰当的描述分子间相互作用,
密度泛函理论 密度泛函理论(DFT)是20世纪60年代建立的并在局域密度近似(LDA)下导出了著名的Koho-Sham(KS)方程。DFT一直是凝聚态物理领域计算电子结构及其特性的有力工具它是一种最常见最成功的研究多电子体系电子结构的量子力学方法。近几年来DFT与分子动力学相结合,在材料设计,合成,模拟计算和评价诸多方面有明显进展,成为计算材料科学的重要基础和核心技术. 特别在量子化学计算领域,1987年以前主要用Hartree-Fock(HF)方法。但近年来,用DFT的工作以指数增加,以致于HF方法应用已相当减少。W.Kohn因提出DFT获得1998年诺贝尔化学奖,已经表明了DFT在计算化学领域的核心作用与应用的广泛性。 密度泛函理论的主要目标就是用电子密度取代波函数作为研究的基本量。因为多电子波函数有3N个变量(N为电子数,每个电子包含三个空间变量),而电子密度仅是三个变量的函数,无论在概念上还是实际上都更方便处理。虽然密度泛函理论的概念起源于Thomas-Fermi模型,但直到Hohenberg-Kohn定理提出之后才有了坚实的理论依据。Hohenberg-Kohn第一定理指出体系的基态能量仅仅是电子密度的泛函。Hohenberg-Kohn第二定理证明了以基态密度为变量,将体系能量最小化之后就得到了基态能量。最初的HK理论只适用于没有磁场存在的基态,虽然现在已经被推广了。最初的Hohenberg-Kohn定理仅仅指出了一一对应关系的存在,但是没有提供任何这种精确的对应关系。正是在这些精确的对应关系中存在着近似(这个理论可以被推广到时间相关领域,从而用来计算激发态的性质。 密度泛函理论最普遍的应用是通过Kohn-Sham方法实现的。在Kohn-Sham DFT的框架中,最难处理的多体问题(由于处在一个外部静电势中的电子相互作用而产生的)被简化成了一个没有相互作用的电子在有效势场中运动的问题。这个有效势场包括了外部势场以及电子间库仑相互作用的影响,例如,交换和相关作用。处理交换相关作用是KS DFT中的难点。目前并没有精确求解交换相关能EXC的方法。最简单的近似求解方法为局域密度近似(LDA近似)。LDA近似使用均匀电子气来计算体系的交换能(均匀电子气的交换能是可以精确求解的),而相关能部分则采用对自由电子气进行拟合的方法来处理。
1、相对于HF方法,DFT方法的优点 2、密度泛函方法:交换泛函和关联泛函 3、绝热近似的基础(内容):核和电子之间的相互运动,近似看做电子不需要时间靠近核的运动 前提:①核的质量大于电子质量,核看成不动,可以考虑分离②不考虑电子从一个态到另一个态的跃迁 4、DFT方法的分类 LDA:slater、 exchange 、VWN condition GGA:Ex B88 PW91 PBE OPTX HCTH,Ec LYP P86 PW91 PBE HCTH LDA和GGA的优缺点: LDA低估了gap,LDA计算晶格常数总是会偏小一些,这样子可以尽可能得到一个电子密度分布均匀的体系,LDA主要Ex就是来自于均匀电子气的交换能,而Ec部分来自于Quantum Monte Carlo计算拟合,对于均匀电子气体系,LDA是理论上严格精确的。 GGA严重低估了CT、里德堡激发的能量,明显低估了gap,GGA优化时电子密度越不均匀的体系,Exc反而越小,体系能量越低。 LDA计算致密结构的能量更接近真实值,而疏松体系的能量都会偏大;GGA相反,疏松结构的能量更接近真实数值,而致密结构则往往偏大 5、Hohenbong-Kohn定理: 一:不计自旋的全同费米子系统的基态,能量是粒子数密度ρ(r)的唯一泛函 二:如果n(r)是体系正确的密度分布,则E[n(r)]是最低能量,即体系的基态能量。 6、DFT的发展方向(前景)---相对于HF方法,DFT方法的优点 DFT方法考虑了电子相关,这会使得过渡态的能量偏低,造成算出来的活化能偏低而且计算氢键的键能也会偏低,而且算起来也快,在计算有机分子的芳香性也不好,dft会过多考虑电子离域,导致计算出来的能量偏低,对于过渡金属、有机生物分子,DFT方法都能很好的处理,这是它比其它方法好的地方。 上个世纪末,很多使用TDDFT算激发能的文章都得到一个相同的结论,就是B3LYP作TDDFT 激发能计算的结果是不可靠的:对不同的分子体系,有的时候跟实验值相当接近,有的时候却差得不得了。因此在做TDDFT激发能计算的时候,应该多试几种泛函,特别是没有实验值。 B3LYP之所以计算TS能量会偏低,主要在于其交换相关势不够准确,特别是在长程区的渐近行为不够好,也正是如此,b3lyp是不可能准确计算氢键. 除一些简单情况(如单-三重态分裂)外,不能普遍用于电子多重态结构的研究,这是密度泛函理论的重要缺陷之一,不解决这个问题,密度泛函理论方法的应用范围受到很大限制。 人们在用密度泛函理论处理多重态分裂问题中针对不同的问题有不同的方法,但各自都有优缺点,没有统一的方法,发表的文章一般只介绍其所用方法的优点,而避开缺点.但DFT的计算量小确实是它的优势,特别是对于大分子体系及磁性材料,半导体材料等性质的研究,所以人们对用DFT计算比较感兴趣. 7、DFT方法选择 非双杂化泛函的最佳选择: 计算碳团簇用B3LYP 计算硼团簇用TPSSh 计算双核金属用PBE、BP86,勿用杂化(see JCTC,8,908) 计算NMR用KT2,M06-L, VSXC, OPBE, PBE0 计算普通价层垂直激发用PBE0(误差约在0.25eV),M06-2X也凑合
密度泛函理论,Density functional theory (DFT) 是一种研究多电子体系电子结构的量子力学方法。密度泛函理 论在物理和化学上都有广泛的应用,特别是用来研究分子和凝聚态的性质,是凝聚态物理和计算化学领域最常用的方法之一。 理论概述 电子结构理论的经典方法,特别是Hartree-Fock 方法和后Hartree-Fock 方法,是基于复杂的多电子波函数的。密度泛函理论的主要目标就是用电子密度取代波函数做为 研究的基本量。因为多电子波函数有
(为电子数,每个电子包含 三个空间变量),而电子密度仅是三个变量的函数,无论在概念上还是实际上都更方便处理。 虽然密度泛函理论的概念起源于Thomas-Fermi 模型,但 直到Hohenberg-Kohn 定理提出之后才有了坚实的理论依据。Hohenberg-Kohn 第一定理指出体系的基态能量仅 仅是电子密度的泛函。 Hohenberg-Kohn 第二定理证明了以基态密度为变量,将体系能量最小化之后就得到了基态能量。 最初的HK理论只适用于没有磁场存在的基态,虽然现在已经被推广了。最初的Hohenberg-Kohn 定理仅仅指出 了一一一对应关系的存在,但是没有提供任何这种精确的对应关系。正是在这些精确的对应关系中存在着近似(这个理论可以被推广到时间相关领域,从而用来计算激发态的性质⑹)。
密度泛函理论最普遍的应用是通过Kohn-Sham 方法实现的。在Kohn-Sham DFT的框架中,最难处理的多体问题(由于处在一个外部静电势中的电子相互作用而产生的)被简化成了一个没有相互作用的电子在有效势场中运动的 问题。这个有效势场包括了外部势场以及电子间库仑相互 作用的影响,例如,交换和相关作用。处理交换相关作用是KS DFT中的难点。目前并没有精确求解交换相关能 的方法。最简单的近似求解方法为局域密度近似(LDA)。LDA近似使用均匀电子气来计算体系的交换能(均匀电子气的交换能是可以精确求解的),而相关能部分则采用对自由电子气进行拟合的方法来处理。 自1970年以来,密度泛函理论在固体物理学的计算中得到广泛的应用。在多数情况下,与其他解决量子力学多体问题的方法相比,采用局域密度近似的密度泛函理论给出了 非常令人满意的结果,同时固态计算相比实验的费用要少。尽管如
1什么是第一性原理? 根据原子核和电子互相作用的原理及其基本运动规律,运用量子力学原理,从具体要求出发,经过一些近似处理后直接求解薛定谔方程的算法,称为第一性原理。广义的第一原理包括两大类,以Hartree-Fock自洽场计算为基础的从头算和密度泛函理论(DFT)计算。 从定义可以看出第一性原理涉及到量子力学、薛定谔方程、Hartree-Fock自洽场、密度泛函理论等许多对我来说很陌生的物理化学定义。因此我通过向师兄请教和上网查资料一点点的了解并学习这些知识。 2第一性原理的作用 以密度泛函理论(DFT)为基础以及在此基础上发展起来的简单而具有一定精度的局域密度近似(LDA)和广义梯度近似(GGA)的第一性原理电子结构计算方法,与传统的解析方法一样,不但能够给出描述体系微观电子特性的物理量如波函数、态密度、费米面、电子间互作用势等,以及在此基础上所得到的体现体系宏观物理特性的参量如结合能、电离能、比热、电导、光电子谱、穆斯堡尔谱等等,而且它还可以帮助人们预言许多新的物理现象和物理规律。密度泛函计算的一些
结果能够与实验直接进行比较,一些应用程序的发展乃至商业软件的发布,导致了基于密度泛函理论的第一原理计算方法的广泛应用。 密度泛函理论(DFT)为第一性原理中的一类,在物理系、化学、材料科学以及其他工程领域中,密度泛函理论(DFT)及其计算已经快速发展成为材料建模模拟的一种“标准工具”。 密度泛函理论可以计算预测固体的晶体结构、晶格参数、能带结构、态密度(DOS)、光学性能、磁性能以及原子集合的总能等等。 3第一性原理怎么用? 目前我所学到的利用第一性原理的软件为Material Studio、V ASP软件。其中Materials Studio(简称MS)是专门为材料科学领域研究者开发的一款可运行在PC上的模拟软件。使化学及材料科学的研究者们能更方便地建立三维结构模型,并对各种晶体、无定型以及高分子材料的性质及相关过程进行深入的研究。模拟的容包括了催化剂、聚合物、固体及表面、晶体与衍射、化学反应等材料和化学研究领域的主要课题。 模块简介 Materials Studio采用了大家非常熟悉的Microsoft标准用户界面,允许用户通过各种控制面板直接对计算参数和计算结果进行设置和分析。目前,Materials Studio软件包括如下功能模块: Materials Visualizer: 提供了搭建分子、晶体及高分子材料结构模型所需要的所有工具,可以操作、观察及分析结构模型,处理图表、表格或文本等形式的数据,并提供软件的基本环境和分析工具以及支持Materials Studio的其他产品。是Materials Studio产品系列的核心模块。 Discover: Materials Studio的分子力学计算引擎。使用多种分子力学和动力学方法,以仔细推导的力场作为基础,可准确地计算出最低能量构型、分子体系的结构和动力学轨迹等。
密度泛函理论及基本应用 凝聚态物理陈阿海2010210602 摘要:本文讨论了密度泛函理论的基本原理和方法,并讨论了玻色子系统中处于谐振子势以及高斯形势垒下的两粒子问题。随着成对的高斯势垒分别向两边移动,粒子数分布由原来被高斯势垒劈裂的两个峰过度到单个峰。 关键词:密度泛函理论,玻色子,谐振势,高斯势垒 Abstract: The basic method of density functional theory is discussed as well as a case of two bosons trapped in a harmonic external potential with Gaussian potential barrier. With the move of a pair of Gaussian potential barrier to the two edges respectively, the distribution of atoms changed gradually from the two peaks divided by the Gaussian potential barrier in the centre of the system at first to single peak again. Key words: density functional theory, boson, harmonic external potential, Gaussian potential barrier 一、引言 密度泛函理论(density functional theory,DFT)是当前研究量子多体系统的重要理论之一,在物理与化学领域均有广泛的应用。DFT的基本出发点是将量子多体系统的性质全部表达为密度的泛函,从而达到简化系统的目的。 密度泛函理论的基本概念最早起源于十九世纪二十年代的Thomas-Fermi模型(TF)[1][2],但其真正意义上的理论开始于六十年代Hohenberg与Kohn的研究,其在1964年发表的著名论文[3]奠定了密度泛函理论的基础。其后,Kohn与Sham在1965年发表的论文将DFT理论推向了实际应用的水平[4],其提出的Kohn-Sham方法成为密度泛函理论的基本应用框架。Kohn凭借其对密度泛函理论的贡献获得了1998年的诺贝尔化学奖。 在其后的几十年间,DFT在多个领域得到了进一步的发展。在量子多体理论的研究中,DFT已发展成为重要的数值计算手段之一,如同量子蒙特卡罗(quantum monte carlo, QMC),密度重整化群(density matrix renormalization group, DMRG),精确对角化(exact diagonalization, ED)等数值方法一样,是当前量子多体系统最主流的数值计算方法之一。为了进一步扩大DFT的应用范围,相继出现了处理含时问题的含时密度泛函理论(time dependent density functional theory, TDDFT)[5-10],处理自旋系统的自旋密度泛函理论(spin density functional theory, SDFT)[11-17],处理有限温度问题的有限温度密度泛函理论(finite temperature density functional theory, FTDFT)[18-20],整合含时密度泛函理论和自旋密度泛函理论的含时自旋密度泛函理论(time-dependent spin-density functional theory, TD-SDFT)[21]等等各种衍生理论和方法。 本文讨论DFT理论的基本原理和方法,进一步利用一个简单的例子讨论DFT的实际应用。在该例子中,主要讨论受谐振势和高斯型势垒作用的一维玻色系统两粒子问题,其中高斯势垒由势阱中央出现并成对向系统两侧移动。主要讨论了该外势作用下系统粒子分布的变化特点。 本文组织如下:第二节中给出了DFT理论的基本框架和特点,主要包括Hohenberg-Kohn 定理,Kohn-Sham方程以及密度泛函理论计算的主要特点;第三节中讨论了零温下受外势作用的一般一维玻色系统的DFT;第四节具体给出了上述所提的两粒子问题的计算结果并作了简单的讨论;最后给出简单的小结。 二、DFT理论的基本框架和特点 基本的DFT理论基于两大基本原理,即Hohenberg-Kohn定理。 定理1指出量子系统的外势与系统的密度分布一一对应(可相差一个常数),或者说,外势是系统密度的泛函,而这种映射是双向的,即系统外势与系统密度分布具有相互确定的一一对应关系。
密度泛函理论-定理介绍 点击查看大图 Density functional theory (DFT) 密度泛函理论是一种研究多电子体系电子结构的量子力学方法。密度泛函理论在物理和化学上都有广泛的应用,特别是用来研究分子和凝聚态的性质,是凝聚态物理和计算化学领域最常用的方法之一。 电子结构理论的经典方法,特别是Hartree-Fock方法和后Hartree-Fock 方法,是基于复杂的多电子波函数的。密度泛函理论的主要目标就是用电子密度取代波函数做为研究的基本量。因为多电子波函数有 3N 个变量(N 为电子数,每个电子包含三个空间变量),而电子密度仅是三个变量的函数,无论在概念上还是实际上都更方便处理。 虽然密度泛函理论的概念起源于Thomas-Fermi模型,但直到Hohenberg-Kohn定理提出之后才有了坚实的理论依据。Hohenberg-Kohn第一定理指出体系的基态能量仅仅是电子密度的泛函。 Hohenberg-Kohn第二定理证明了以基态密度为变量,将体系能量最小化之后就得到了基态能量。 最初的HK理论只适用于没有磁场存在的基态,虽然现在已经被推广了。最初的Hohenberg-Kohn定理仅仅指出了一一对应关系的存在,但是没有提供任何这种精确的对应关系。正是在这些精确的对应关系中存在着近似(这个理论可以被推广到时间相关领域,从而用来计算激发态的性质[6])。 密度泛函理论最普遍的应用是通过Kohn-Sham方法实现的。在Kohn-Sham DFT的框架中,最难处理的多体问题(由于处在一个外部静电势中的电子相互作用而产生的)被简化成了一个没有相互作用的电子在有效势场中运动的问题。这个有效势场包括了外部势场以及电子间库仑相互作用的影响,例如,交换和相关作用。处理交换相关作用是KS DFT中的难点。目前并没有精确求解交换相关能 EXC 的方法。最简单的近似求解方法为局域密度近似(LDA)。LDA近似使用均匀电子气来计算体系的交换能(均匀电子气的交换能是可以精确求解的),而相关能部分则采用对自由电子气进行拟合的方法来处理。 GW近似用于计算多体系统中的自能。利用Green函数G与含屏蔽的相互作用W 对体系自能做展开: GW近似就是截取该展开式的首项:
μ—η2:η2—[Cu2O2]2+和Bis(μ-OXO)—[Cu2O2]2+ 的密度泛函理论研究 李勇,张颂富,董娴,陈卓 (贵州民族学院化学与环境科学学院,贵阳550004;2.贵州师范大学理学院,贵阳550001) 摘要:酪氨酸酶是一种氧化酪氨酸残基为对苯醌的生物活性酶,其活性部位含有双铜核心并参与氧化还原反应。实验证明这两个铜核心易与氧气结合生成两种异构体μ—η2:η2—Cu2O2(Ⅱ)和bis(μ-OXO)-Cu2O2(Ⅲ),且它们具有不同的电子结构和化学性质。用量子化学密度泛函理论对乙(撑)二胺和乙腈配合的μ—η2:η2—Cu2O2(Ⅱ)和bis(μ-OXO)-Cu2O2(Ⅲ)进行了理论研究,结果表明该两种异构体能量差别较小,相互转化的势垒较低。μ—η2:η2—Cu2O2(Ⅱ)中的Cu-O主要是离子键,Cu2O2呈“Z”构型,而bis(μ-OXO)-Cu2O2(Ⅲ)中的Cu-O主要是共价键,呈“V”构型,在氧化还原反应中具有更强的亲电子能力。关键词:DFT(密度泛函理论);Cu2O2;可逆反应;互变异构;化学平衡 中图分类号:O643.12 文献标识码:A 1 引言 近年来,异构体bis(μ-OXO)复合物和[Mn(μ-O2)]n+(M=Cu,Mn,Ni,Co,Fe和Pt)核由于它们在生物学和仿生氧化上的关联被广泛研究。各种金属酶普遍包含一个或多个金属原子,它们激活分子氧氧化有机基质。酪氨酸酶是种生物活性酶,其中[Cu2(μ-O2)]2+核心被认为能氧化酪氨酸残基为对苯醌,如同颗粒性甲烷单加氧酶在异质沸石基系统中生成甲烷羟化酶。双核铜氧酶可以因磁相互作用在两个Cu(Ⅱ)离子间被分成两部分,例如反铁磁性的成对部分和非成对部分。就是说,漆酶包含一对反磁铁性的铜离子,可以看成是双电子氧化剂。因此探索新生铜氧的反应关系结构是很有意义的。为了效仿这种自然系统,一些合成模式以双核铜血蓝蛋白为依据,其中每个铜被三个组氨酸残基以减化和氧化的形式绑扎,另一些依靠空间的阻碍,三齿配合物。然而,有人提出,每个铜齿氮结扎应该足以稳定2:1的Cu/ O2中间体,因为在氧合血蓝蛋白和所有结构表征模型2:1的Cu/ O2中间体采用齿配体,各铜配位层中的一个氮原子是弱相关联,并且被认为是沿铜中心的杨 - 特勒轴线定位。
密度泛函 密度泛函理论, Density functional theory (DFT) 是一种研究多电子体系电子结构的量子力学方法。密度泛函理论在物理和化学上都有广泛的应用,特别是用来研究分子和凝聚态的性质,是凝聚态物理和计算化学领域最常用的方法之一。 目录 简介 密度泛函理论(Density Functional Theory,DFT),是基于量子力学和玻恩-奥本海默绝热近似的从头算方法中的一类解法,与量子化学中基于分子轨道理论发展而来的众多通过构造多电子体系波函数的方法(如Hartree-Fock类方法)不同,这一方法构建在一个定理的基础上:体系的基态唯一的决定于电子密度的分布(Hohenberg-Kohn定理),从而使得我们可以采用最优化理论,通过KS-SCF自洽迭代求解单电子多体薛定谔方程来获得电子密度分布,这一操作减少了自由变量的数量,减小了体系物理量振荡程度,并提高了收敛速度,并易于通过应用HF定理等手段,与分子动力学模拟方法结合,构成从头算的分子动力学方法。这一方法在早期通过与金属电子论、周期性边界条件及能带论的结合,在金属、半导体等固体材料的模拟中取得了较大的成功,后来被推广到其它若干领域。目前常见的基于DFT的商业软件有:VASP,CASTEP等。 Hohenberg-Kohn第二定理 密度泛函理论中的另一条重要定理是Hohenberg-Kohn第二定理证,它证明了以基态密度为变量,将体系能量最小化之后就得到了基态能量。 最初的HK理论只适用于没有磁场存在的基态,虽然现在已经被推广了。最初的Hohenberg-Kohn定理仅仅指出了一一对应关系的存在,但是没有提供任何这种精确的对应关系。正是在这些精确的对应关系中存在着近似(这个理论可以被推广到时间相关领域,从而用来计算激发态的性质[6])。
密度泛函理论简介 密度泛涵理论最初来源于对下面这个问题的考虑: 在量子化学从头算中,对于一个N电子体系,N电子波函数依赖于3N个空间变量及N个自旋变量共4N个变量,我们是否能其它相对简单的变量来替换这4N个变量以达到简化计算的目的,如用体系的电子密度?因为,对于波函数实 验上无法准确测定,而电子密度却可以,电子密度同波函数模的平方相联系.另一方面,对于 依赖4N个变量的波函数,将随着体系变大电子数增多使计算变得越来越困难,而体系的哈密 顿只不过由单电子和双电子算符组成,同时只跟体系中的单个电子和双电子的信息有关,因 此波函数中4N个变量已经包含了多余的信息,对我们的计算目的而言.因此,以电子密度为变量,Thomas-Fermi Model作了最初的尝试,将能量表示为密度的泛函,这里有个问题要注意的 是泛函和复合函数的区别.TFM虽然是一个很粗糙的模型,但是它的意义非常重要,因为它将 电子动能第一次明确地以电子密度形式表示.至此,说简单些,密度泛函方法就是以体系的电 子密度为变量的方法. 随后,Hohenberg-Kohn定理证明了external potentail是密度的唯一泛函,多电子体系的 基态也是电子密度的唯一泛函.因此,对于多电子体系非简态基态而言有一基态电子密度相 对应,,正是这个基态电子密度也决定了体系的基态的其它性质,寻找基态的电子密度同样利 用变分方法.有关这个定理的内容可以参考其它资料. 在此定理的基础上,Kohn and Sham引入了"无相互作用参考系统"的概念,这个思想和传 统的从头算不同,我们推导的HF方程是建立在真实的系统基础上的,而无相互作用参考系统 是不存在的,只是KS为计算真实体系的设立的一个参照系统,它和真实系统的联系就在于有 相同的电子密度.因此,我们也可以看出,DFT能获Nobel Prize也是完全在于它是一个全新的 创造性的思想.这个无相互作用系统中,粒子间无相互作用,它的哈密顿算符就只有两项,动 能算符和势能算符,这个形式和HF方法的形式比起来就简单多了,同HF方程一样,根据单电子近似也得到了KS单电子算符.接下来就是将这个参照系统同真实系统联系起来.HF方法完全 忽略了相关能的计算,在DFT中,这部分能量考虑了进去,因此从原理上讲,Kohn-Sham方法是 严格的,未作任何近似,但是同交换相关能相联系的交换相关势的形式却是无法确定的,因此 DFT的中心问题更是寻找更好的泛函形式.