
离子色谱非抑制法测定草甘膦中的胺及异丙胺
摘要:本文采用离子色谱非抑制型电导快速测定草甘膦中的胺及异丙胺的含量。采用Ionpac SCG1+ SCS1阴离子分析柱分离,以电导为检测器。用去离子水稀释1000倍后直接进样。实验结果表明,在一定的色谱条件下,胺及异丙胺具体很好的线性,和重现性,及较低的检出限。最低检出量0.17ug/l和58.5ug/l。样品的平均加标回收率为90-103%。
关键词:离子色谱;农药;草甘膦;胺;异丙胺;非抑制
引言
草甘膦为内吸传导型广谱灭生性除草剂。主要抑制物体内烯醇丙酮基莽草素磷酸合成酶,从而抑制莽草素向苯丙氨酸、酷氨酸及色氨酸的转化,使蛋白质的合成受到干扰导致植物死亡。由于草甘膦原药的水溶性差,必须加工配制成铵盐、异丙胺盐等草甘膦盐类才能溶于水使用。而农业部对按草甘膦铵盐和草甘膦异胺盐名称登记的产品,标示的有效成分是指草甘膦铵盐的含量。因而通过测定成品草甘膦铵盐及草甘膦异丙胺盐中铵以及异丙胺的含量,从而可以换算为相应盐的含量。这对于生产草甘膦农药的厂家的质量控制以及反应工艺过程的控制是非常重要的。https://www.doczj.com/doc/ae553821.html,/Shop/Product/Product_193965.shtml
以往文献中报道对于草甘膦铵盐和异丙胺盐含量的测定,一般是先用特定的方法测得草甘膦的含量,再转换成草甘膦铵盐和草甘膦异丙胺盐的含量。对于草甘膦含量的测定,已有薄层层析法[1]、分光光度法[2]、气相色谱法[3-4]、高效液相色谱法[5-7]等方法,其中对采用气相色谱或高效液相色谱测定其含量的研究较多。由于草甘膦极性很强,无论是采用气相还是高效液相色谱进行测定,都需要进行衍生,但衍生化需要用到对人体可能有潜在危害的试剂,同时测定步骤复杂。也有人对采用分光光度法测定草甘膦进行了研究,但容易受到一些共存的化合物和离子的干扰,从而影响结果的准确性。国内已有报道采用离子色谱法测定草甘膦,从而间接得到草甘膦异丙胺盐的含量。但在实际的市场监测中,单单测定草甘膦的含量并无法知道此草甘膦水剂是草甘膦铵盐亦或者草甘膦异丙铵盐。因此上述方法在实际应用中都有一定的局限性。中国色谱网https://www.doczj.com/doc/ae553821.html,
在本文中建立了采用离子色谱非抑制型电导快速测定草甘膦铵盐以草甘膦异丙胺盐水剂中的铵以及异丙胺的含量,可做为草甘膦含量以及种类确认的手段。方法简便,测定结果的值稳定性高,干扰小,灵敏度好。
1.实验部分
1.1 仪器
Dionex ICS 2000 离子色谱仪(美国Dionex公司);DS6电导检测器;Chromeleon 6.8 色谱工作站。
1.2 试剂
所有试剂均为分析纯。异丙胺以及氯化铵由杭州惠普科技有限公司提供。标准溶液均由1000mg/L的贮备液(置于4℃冰箱中)稀释配制,溶液均用18.2MΩ.cm 的二次去离子水配制。样品为草甘膦农药生产厂家提供。
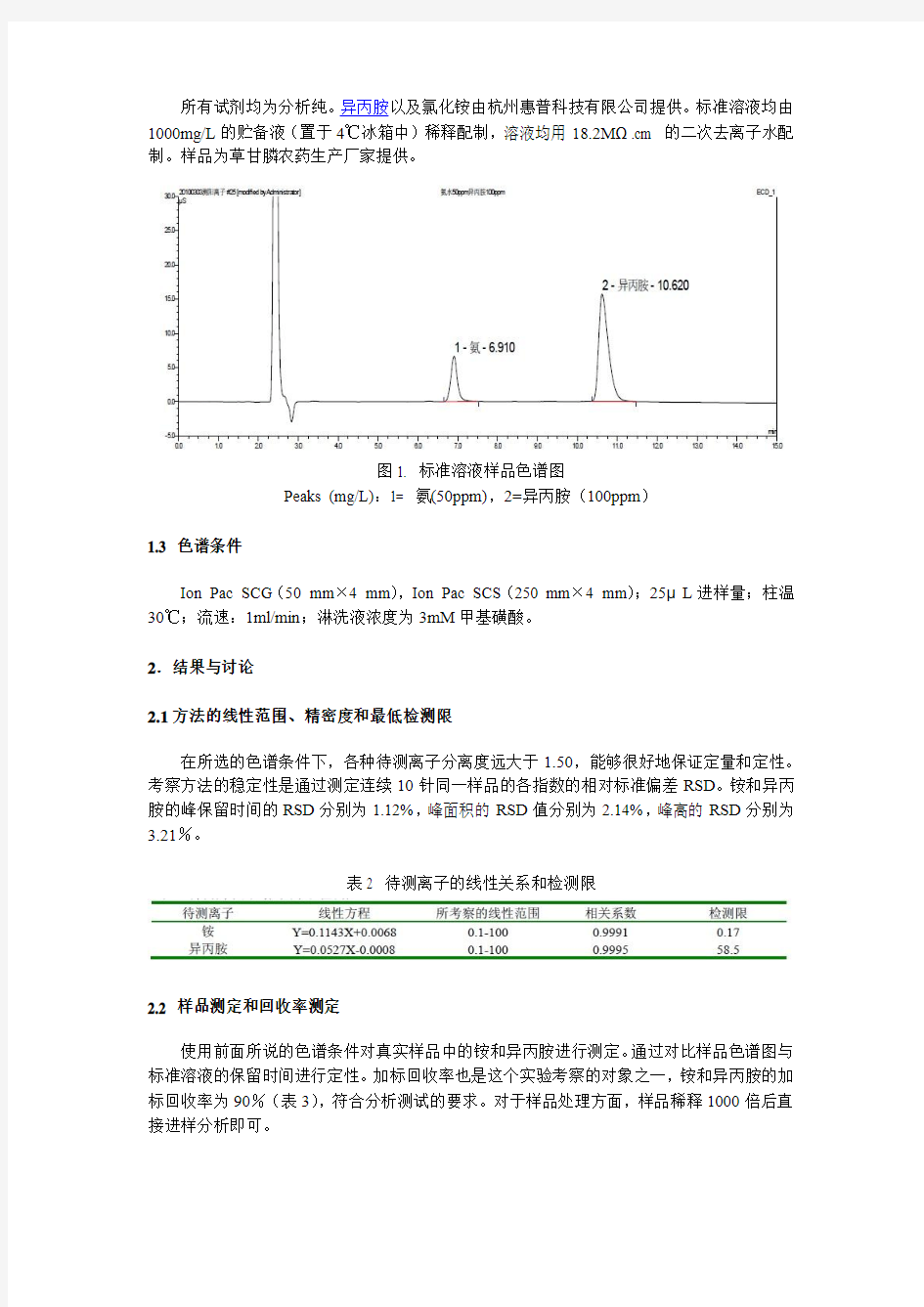
图1. 标准溶液样品色谱图
Peaks (mg/L):1= 氨(50ppm),2=异丙胺(100ppm)
1.3 色谱条件
Ion Pac SCG(50 mm×4 mm),Ion Pac SCS(250 mm×4 mm);25μL进样量;柱温30℃;流速:1ml/min;淋洗液浓度为3mM甲基磺酸。
2.结果与讨论
2.1方法的线性范围、精密度和最低检测限
在所选的色谱条件下,各种待测离子分离度远大于1.50,能够很好地保证定量和定性。考察方法的稳定性是通过测定连续10针同一样品的各指数的相对标准偏差RSD。铵和异丙胺的峰保留时间的RSD分别为1.12%,峰面积的RSD值分别为2.14%,峰高的RSD分别为3.21%。
表2 待测离子的线性关系和检测限
2.2 样品测定和回收率测定
使用前面所说的色谱条件对真实样品中的铵和异丙胺进行测定。通过对比样品色谱图与标准溶液的保留时间进行定性。加标回收率也是这个实验考察的对象之一,铵和异丙胺的加标回收率为90%(表3),符合分析测试的要求。对于样品处理方面,样品稀释1000倍后直接进样分析即可。
表3 待测离子的加标回收率
图2 样品1稀释100倍的色谱图
Peaks:1= 铵,2=异丙胺
图3 样品2稀释1000倍的色谱图
Peaks:1= 铵,2=异丙胺
3 结论
本文提出的方法以离子色谱非抑制电导法测定草甘膦铵盐以及草甘膦异丙胺盐中的铵和异丙胺,灵敏度高,方法简便,分离时间短,可广泛应用于生产过程中的产品质量控制及市场商品检验。
参考文献
[1] Melvin L Rueppel. Metabolism and Degredation of Glyphosate in Soiland Water [J]. Agric.
food chem., 1977, 25 (3): 517–518.
[2] 李水清, 叶森林. 草甘膦在土壤中残留动态研究[J]. 长江大学学报(自科版), 2006, 3 (1): 123–125.
[3] 楼正云, 朱国念, 吴慧明. 池塘水中草甘膦残留检测方法的研究[J]. 宁波高等专科学校学报, 2006, 13 (增刊): 141–144.
[4] 裴茂清, 赖杰. 草甘膦的定性定量分析方法[J]. 广东公安科技,2004, (1): 14–15.
[5] Waters公司北京实验室. 液相色谱法测定饮用水中的草甘膦[J].环境化学, 1995, 14 (6): 554–555.
[6] 候占伟, 王朝虹. 高效液相色谱法测定水中的草甘膦[J]. 刑事技术, 2003, (6): 9–10.
[7] 胡宝祥, 莫卫民, 孙楠. ELSD–HPLC 联用测定制剂中草甘膦的含量[J]. 农药, 2005, 44
(8): 365–367.
[8]武中平.离子色谱法测定草甘膦异丙胺盐的含量.现代农药,2008,7(4): 31-35
上海百贺仪器科技有限公司提供www.southhk.cn 紫外分光光度法测定蛋白质含量 摘要: 考马斯亮兰G250与蛋白质结合,在0-1000ug/ml范围内,于波长595nm 处的吸光度与蛋白质含量成正比,可用于蛋白质含量的测定。考马斯亮兰G250 与蛋白质结合迅速,结合产物在室温下10分钟内较为稳定,是一种较好的蛋白 质定量测定方法。 1.实验部分 1.1仪器与试剂: Labtech UV POWER紫外分光光度计;玻璃比色皿一套;考马斯亮蓝G250; 牛血清蛋白;超纯水。 1.2试液的制备: 牛血清蛋白标准溶液(1000ug/ml)的制备称取100mg牛血清蛋白置100ml 容量瓶中,加入超纯水溶解并定容。 考马斯亮兰G250试剂称取100mg考马斯亮兰G250,溶于50ml95%的乙 醇后,加入120ml85%的磷酸,用水稀释至1升。 2.结果与讨论 2.1校正曲线的绘制 准确吸取1000ug/ml牛血清蛋白标准溶液0.0、0.02、0.04、0.06、0.08、0.1ml 分别加入到6只10ml试管中,然后用超纯水补充到0.1ml,各试管分别加入5ml 考马斯亮兰G250试剂,混合均匀后,即可依次在595nm处测定吸光度。以浓度 为横坐标,吸光度为纵坐标绘制校正曲线如下图,校正曲线方程为 A=0.613556C+0.001008,R=0.9994。
上海百贺仪器科技有限公司www.southhk.cn 2.2精密度 配制0.6mg/ml牛血清蛋白的考马斯亮兰溶液连续进样6次,得到吸光度的 相对标准偏差。 表1精密度测定结果 次数123456RSD% A0.26260.26220.26200.26280.26290.26260.13 2.3稳定性 取1mg/ml牛血清蛋白标准溶液每十分钟测定一次,50分钟内的吸光度变化 如下表2。 表2稳定度测定结果 时间(min)A1A2A3A平均 00.55110.55230.55160.5517 100.52040.51840.51680.5185 200.49100.49010.49030.4905 300.47650.47160.47210.4734 400.45240.44750.44400.4480 500.39820.39350.40310.3983 3.结论 该方法测定快速、简便,干扰物少,是目前灵敏度较高的蛋白质含量测定 的紫外分光光度法。
离子色谱法都有哪些干扰及解决办法 一般来说,离子色谱仪在操作过程中比较常见的故障有如下几种,化学实验员有必要学习下离子色谱仪一些简单的维护和操作。(本栏问题的维护仅供参考,实际维护中配件如果与此有差异,均可以参考下列操作) 一、由流动相到泵之间的管路中有气泡,怎么排除? 排除方法如下:先将与泵相连的塑料流路接头拧下来,用洗耳球吸满去离子水,从与泵段相连的流路管中注入,将流路管中的气泡排除干净。然后再将流动相瓶(一般为去离子水瓶)抬高,再将流路接头与泵连接好。启动泵,打开泵内排气阀选钮,将泵内气泡排除干净,一般观察为流出液比较均匀,再将泵排气阀拧紧。(注意:此项操作时,整个流路是与色谱柱断开的) 二、泵单向阀堵塞会有哪些现象?怎么操作? 在如果泵单向阀上粘上了微生物造成堵塞会造成泵吸液不上,最明显的现象是,在废液管没有流液或启动泵时没有液体流出或溶液流出速度很慢。 单向阀如果堵塞了,我们需要对其进行清洗,清洗方法如下: 先将流路接头和接头1全部拧下,再将左侧接头2拧下,用镊子将两单向阀取出放入50ml 烧杯中(取出时注意观察单向阀上箭头方向,安装时方向必须与此相同),加入少量无水乙醇刚好盖过两单向阀。再放入超声波清洗器中清洗30-60min。再用水将单向阀清洗干净后按拆卸方向逆向安装即可。(注意:接头不要拧的太紧,以免造成螺丝纹受损) 三、抑制器电流无显示,怎么判断问题?怎么操作? a、先要了解抑制器的结构,抑制器的结构见图3: b、抑制器电路的检测:用螺丝刀将抑制器盒四角4颗固定螺丝拧下,就可以看到如图3所示的抑制器图。将抑制器两边的电极线取下,注意两边电极线的颜色不一致(一般红色线接在右边,左边为灰色或黑色线),再用配件工具箱中的模拟电阻(100欧姆)连接两电极线,再顺时针打开并调节控制面板上的电流旋钮,观察触摸屏上电流显示数字的变化。若变化规则,从45-100或105mA可调,说明抑制器电路运行正常,那么抑制器电流显示不正常的原因来源于两电极线连接到抑制器上时接触不良所致。若无变化,一直显示为0,则说明抑制器电路已经被击穿,需要重新更换电路或对其进行维护。 c、抑制器上四方流路接头的翻边和连接:从电导池出口再次进入抑制器时其接头的连接需要翻边的操作。 d、抑制器接头的连接和漏液问题的维护:抑制器上的接头主要有3个,一个接色谱柱出口(PEEK锥形接头),另外两个接电导池入口(PEEK锥形接头) 和出口(四方流路接头),接头接的位置均在抑制器上有所标明,连接的时候注意不要弄错了。另外,接头的连接均是宜松不宜紧,以不泄漏为宜。接色谱柱出口端接头拧的太紧,会造成系统压力增大。电导池出口的四方流路接头拧得太紧会造成溶液不流通电导池,更有甚者会造成抑制器的离子交换膜破裂而损坏抑制器。
实验一邻二氮菲分光光度法测定微量铁 实验目的和要求 1.掌握紫外可见分光光度计的基本操作; 2.掌握邻二氮菲分光光度法测定微量铁的原理和方法; 3.掌握吸收曲线绘制及最大吸收波长选择; 4.掌握标准曲线绘制及应用。 实验原理 邻二氮菲(1,10—邻二氮杂菲)是一种有机配位剂,可与Fe2+形成红色配位离子: Fe2++3 N N N N 3 Fe 2+ 在pH=3~9范围内,该反应能够迅速完成,生成的红色配位离子在510nm波长附近有一吸收峰,摩尔吸收系数为1.1×10-4,反应十分灵敏,Fe2+ 浓度与吸光度符合光吸收定律,适合于微量铁的测定。 实验中,老师我们又见面了采用pH=4.5~5的缓冲溶液保持标准系列溶液及样品溶液的酸度;采用盐酸羟胺还原标准储备液及样品溶液中的Fe3+并防止测定过程中Fe2+被空气氧化。 实验仪器与试剂 1.752S型分光光度计 2.标准铁储备溶液(1.00×10-3mol/L) 3.邻二氮菲溶液(0.15%,新鲜配制) 4.盐酸羟胺溶液(10%,新鲜配制) 5.NaAC缓冲溶液 6.50ml容量瓶7个 7.1cm玻璃比色皿2个 8.铁样品溶液 实验步骤 1.标准系列溶液及样品溶液配制,按照下表配制铁标准系列溶液及样品溶液。
2.吸收曲线绘制用1cm比色皿,以1号溶液作为参比溶液,测定4号溶液在各个波长处的吸光度,绘制吸收曲线,并找出最大吸收波长。 3.标准曲线制作
在选定最大吸收波长处,用1cm 比色皿,以1号溶液作为参比溶液,分别测定2至7号溶液的吸光度,平行测定3次,计算吸光度平均值,绘制标准曲线。 实验数据处理 1、 样品中铁的计算 2.50 50.00 C C X ? =读取值 Cx=4.65×10-5 ×50.00/2.50=9.30×10-4 mol/L 2、 摩尔吸光系数计算 在标准曲线的直线部分选择量两点,读取对应的坐标值,计算邻二氮菲配位物在最大吸收波长出的摩尔吸光系数: 1 21 2c -c A A ε-= ε=(0.460-0.233)/(0.00006-0.00004)=2.00×10-5 7 样品溶液 4.65×10-5 mol/ml
41%草甘膦异丙胺盐水剂防除桑园杂草田间药效试验 摘要41%草甘膦异丙胺盐水剂防除桑园杂草田间药效试验结果表明,该药剂施用剂量不低于 3.0L/hm2,即有效成分 1.230kg/hm2,采用二次稀释法,对水750L/hm2,在杂草旺盛生长时期进行杂草茎叶定向均匀喷雾,对桑园杂草防效好,持效期长,对桑树安全,无药害。 关键词41%草甘膦异丙胺盐水剂;桑园杂草;防效 高含量草甘膦除草剂是今后草甘膦类除草剂发展及应用的主要方向。为明确江苏丰山集团有限公司研制的41%草甘膦异丙胺盐水剂对桑园杂草的防效,特进行了本试验。 1材料与方法 1.1试验田概况 试验田设在金寨县白塔畈乡楼冲村一农户承包田,面积780m2,土壤为黄棕壤,土壤质地为中层耕种麻石土,有机质含量16.0g/kg,pH值5.2,施肥、管理与当地生产水平一致。试验田桑树为胡桑,1998年栽植,株行距0.5m×2.0m,长势较好。试验时桑树呈光拳状,部分夏伐处开始吐露新叶;桑园内的杂草正处于旺盛生长时期,多数杂草处于八至十叶期。试验时土壤相对含水量80%左右。 1.2试验对象 药前进行桑园田间杂草密度调查,平均密度为245株/m2左右,其中狗尾草(Setaria viridis(L.)Beauv)约占36%,碎米莎草(Cyperus iria L.)约占23%,牛筋草(Eleusine indica(L.)Gaertn)约占14%,南苜蓿(Medicago hispida Gaertn)约占12%,其他少量杂草包括小飞蓬、铁苋菜、酸模叶蓼、鸡眼草、萹蓄、苍耳、反枝苋、粟米草、稗草、马唐及其他莎草等,约占15%。 1.3供试药剂
紫外-可见分光光度法测有色溶液最大吸收波波长 一、实验目的 1.学习紫外-可见分光光度法的原理; 2.掌握紫外-可见分光光度法测定的实验技术; 3.了解掌握U-3010型紫外-可见分光光度仪的构造及使用方法。 二、实验原理 1.紫外-可见吸收光谱法(称紫外-可见分光光度法)以溶液中物质的分子或离 子对紫外和可见光谱区辐射能的选择性吸收为基础而建立起来的一类分析法。根据最大吸收波长可做定性分析;根据朗伯-比尔定律(标准曲线法和标准加入法)可做定量分析。紫外-可见分光光度法定性分析原理:根据吸收曲线中吸收峰的数目、位置、相对强度以及吸收峰的形状进行定性分析。 2.紫外-可见分光光度法定量分析原理,根据朗伯-比耳定律:A=εbc,当入 射光波长λ及光程b一定时,在一定浓度范围内,有色物质的吸光度A与该物质的浓度c成正比。定量分析常用的方法是标准曲线法即只要绘出以吸光度A为纵坐标,浓度c为横坐标的标准曲线,测出试液的吸光度,就可以由标准曲线查得对应的浓度值,即未知样的含量。 3.仪器由五个部分组成:即光源、单色器、吸收池、检测器和信号显示记录装 置。 三、仪器与试剂 日立U-3010型紫外-可见分光光度仪;吸量管;乙醇;待测溶液;烧杯等。 四、实验步骤 1.接通电源,启动计算机,打开主机电源开关,启动工作站并初始化仪器,预 热半小时。 2.在工作接口上选择测量项目为光谱扫描,设置扫描参数(起点:650nm,终 点:250nm,速度:中,间隔:1.0nm,单次扫描) 3.将两个均装有无水乙醇的1cm石英比色皿放入测量池中,进行基线扫描。 4.基线做好后,按下面的顺序进行操作:做Baseline→换样(换上待测样品置 于Sample池)→进入Analysis Method对相关的参数进行设定→Sample命名→Ready→Measure进行测量,寻找待测溶液的最大吸收波长,再在最大吸收波长处分别测定待测溶液的吸光度。
离子色谱仪技术参数 1应用范围:适用于样品中阴阳离子、有机酸及有机胺类物质的分析,满足饮用水GB5750中阴阳离子检测标准,同时能跟ICP-MS联用用于元素的形态学分析。 2技术要求 2.1离子色谱系统,包括淋洗液瓶,泵,内置电动六通阀,保护柱,分析柱,阴阳离子抑制器和电导检测器。 2.2所有的离子色谱流路均标配采用PEEK材质,须包括分析泵本身及分析泵后至六通阀、色谱柱、抑制器、检测器之间的所有管路。 2.3▲泵:高性能/低脉冲双柱塞泵,采用化学惰性的非金属无阻尼泵头,PEEK管路。适合于pH为0~14的淋洗液及反相有机溶剂。流速范围:0.00-5.00 mL/min(可选配0.00-10.00 mL/min),最大压力:5000psi,流速最大误差<0.1%,流量精密度:<0.1%,压力脉冲:小于系统压力的1.0%,可升级为二元高压梯度淋洗系统。 2.5▲高压梯度重复性偏差限:<0.2%,高压梯度误差限: <0.15%。 2.6电导检测器:与自动电解连续再生微膜抑制器联用,降低系统背景,提高信噪比。 2.6.1类型:数字信号控制处理器温度补偿功能: 2.6.2须具有温度补偿功能。
2.6.3电导池体积:<1.0 μL,全程信号输出范围:0-10000μS,检测器分辨率:≤0.0047 nS/cm。 2.6.4▲检测器耐受最大压力:≥8Mpa。 2.6.5电导池电极材料:钝化316不锈钢。 2.6.6电导池体材料:化学惰性聚合材料。 2.6.7线性:1%。 2.7▲电解自动再生离子交换抑制器:用电解水自动产生H+和OH ̄进行离子交换中和反应的抑制技术。 2.7.1阴离子自动电解连续再生微膜抑制器,无需外加硫酸进行轮流再生。 2.7.2▲阳离子自动电解连续再生微膜抑制器,具备连续电解再生抑制功能。 2.8▲色谱分析柱:高效高容量分离柱(250*4 mm)及相应的保护柱(50*4mm)组成,色谱柱须采用聚合物基质,耐受pH 0-14的工作范围,可耐受3000 psi以上压力,100%兼容反相试剂,可使用强酸强碱淋洗液。 2.8.1可以使用自动电解连续再生微膜抑制器或化学连续再生微膜抑制器的高效高容量阴离子分离柱,色谱柱须采用聚合物基质,耐受pH 0-14的工作范围,柱交换量220 μeq/根以上,可一次进样完成阴离子和溴酸根的分析。 2.8.2▲可以使用自动电解连续再生微膜抑制器或化学连续再生微膜抑制器的高效高容量阳离子分离柱及保护柱,色谱柱
专业项目课程课例 项目十二分光光度法测定水中铁离子含量 一、项目名称:分光光度法测定水中铁离子含量 二、项目背景分析 课程目标:本课程是培养分析化学操作技能和操作方法的一门专业实践课,以定量分析的基本理论为基础,以实验强化理论,以期提高化工工作者的分析操作能力。 功能定位:在定量分析中我们常常用到分光光度分析法,它具有操作简便、快速、准确等优点,在工农业生产和科学研究中具有很大的实用价值。是仪器分析的基础实验,也是一种重要的定量分析方法。分光光度法测定水中铁离子含量的测定项目综合训练了学生分光光度计使用、系列标准溶液配制、标准曲线绘制等多个技能。 学生能力:学生通过相关基础学科的学习已经具备了相应的化学知识和定量分析知识,也具备一定的独立操作和思维能力。 项目实施条件:该项目是仪器分析的基础实验,一般中职学校具备相关的实训实习条件,学生有条件完成相应的实习任务。 三、教学目标 1、了解721可见分光光度计的构造 2、了解分光光度法测定原理 3、掌握721可见分光光度计的操作方法 4、掌握分光光度法测定分析原始记录的设计 5、掌握分光光度法测定分析报告的设计 6、掌握分光光度法测定水中铁离子含量的测定方法 7、掌握分光光度法测定水中铁离子含量的分析原始记录和分析报告的填写 四、工作任务 1
2 五、参考方案 参考方案一 1、邻二氮杂菲-Fe 2+ 吸收曲线的绘制 用吸量管吸取铁标准溶液(20μg/mL )0.00、2.00、4.00mL ,分别放入三个50mL 容量瓶中,加入1mL 10%盐酸羟胺溶液,2mL 0.1%邻二氮杂菲溶液和5mL HAc-NaAc 缓冲溶液,加水稀释至刻度,充分摇匀。放置10min ,用3cm 比色皿,以试剂空白(即在0.0mL 铁标准溶液中加入相同试剂)为参比溶液,在440~560nm 波长范围内,每隔20~40nm 测一次吸光度,在最大吸收波长附近,每隔5~10nm 测一次吸光度。在坐标纸上,以波长λ为横坐标,吸光度A 为纵坐标,绘制A 和λ关系的吸收曲线。从吸收曲线上选择测定Fe 的适宜波长,一般选用最大吸收波长λmax 。 2、标准曲线的制作 用吸量管分别移取铁标准溶液(20μg/mL )0.00、2.00、4.00、6.00、8.00、10.00mL ,分别放入6个50mL 容量瓶中,分别依次加入1.00mL 10%盐酸羟胺溶液,稍摇动;加入2.00mL 0.1%邻二氮杂菲溶液及5.00mL HAc-NaAc 缓冲溶液,加水稀释至刻度,充分摇匀。放置10min ,用1cm 比色皿,以试剂空白(即在0.00mL 铁标准溶液中加入相同试剂)为参比溶液,选择λmax 为测定波长,测量各溶液的吸光度。在坐标纸上,以含铁量为横坐标,吸光度A 为纵坐标,绘制标准曲线。 3、水样中铁含量的测定 取三个50mL 容量瓶,分别加入5.00mL (或10.00mL 铁含量以在标准曲线范围内为合适)未知试样溶液,按实验步骤2的方法显色后,在λmax 波长处,用1cm 比色皿,以试剂空白为参比溶液,平行
6种方法测定蛋白质含量 一、微量凯氏(kjeldahl)定氮法 样品与浓硫酸共热。含氮有机物即分解产生氨(消化),氨又与硫酸作用,变成硫酸氨。经强碱碱化使之分解放出氨,借蒸汽将氨蒸至酸液中,根据此酸液被中和的程度可计算得样品之氮含量。若以甘氨酸为例,其反应式如下:nh2ch2cooh+3h2so4——2co2+3so2+4h2o+nh3 (1) 2nh3+h2so4——(nh4)2so4 (2) (nh4)2so4+2naoh——2h2o+na2so4+2nh3 (3) 反应(1)、(2)在凯氏瓶内完成,反应(3)在凯氏蒸馏装置中进行。 为了加速消化,可以加入cuso4作催化剂,k2so4以提高溶液的沸点。收集氨可用硼酸溶液,滴定则用强酸。实验和计算方法这里从略。 计算所得结果为样品总氮量,如欲求得样品中蛋白含量,应将总氮量减去非蛋白 氮即得。如欲进一步求得样品中蛋白质的含量,即用样品中蛋白氮乘以6.25即得。 二、双缩脲法(biuret法) (一)实验原理 双缩脲(nh3conhconh3)是两个分子脲经180℃左右加热,放出一个分子氨后得到的产物。在强碱性溶液中,双缩脲与cuso4形成紫色络合物,称为双缩脲反应。凡具有两个酰胺基或两个直接连接的肽键,或能过一个中间碳原子相连的肽键,这类化合物都有双缩脲反应。 紫色络合物颜色的深浅与蛋白质浓度成正比,而与蛋白质分子量及氨基酸成分无关,故可用来测定蛋白质含量。测定范围为1-10mg蛋白质。干扰这一测定的物质主要有:硫酸铵、tris缓冲液和某些氨基酸等。 此法的优点是较快速,不同的蛋白质产生颜色的深浅相近,以及干扰物质少。主要的缺点是灵敏度差。因此双缩脲法常用于需要快速,但并不需要十分精确的蛋白质测定。 (二)试剂与器材 1. 试剂: (1)标准蛋白质溶液:用标准的结晶牛血清清蛋白(bsa)或标准酪蛋白,配制成10mg/ml的标准蛋白溶液,可用bsa浓度1mg/ml的a280为0.66来校正
HZHJSZ0092 水质肼的测定对二甲氨基苯甲醛分光光度法 HZ-HJ-SZ-0092 水质对二甲氨基苯甲醛分光光度法 1 范围 本方法规定了测定水中肼的对二甲氨基苯甲醛分光光度法 试料体积1~10mL±?·?·¨?ì2a?Tò??????a0.002mg/L 更高浓度的样品 氨基脲脲素分别高达20200mg/L以上时干扰测定 NO2ˉ 大于1mg/L时产生干扰 肼与对二甲基苯甲醛作用于波长458nm 处进行分光光度测定 本方法所有试剂均为符合国家标准或专业标准的分析纯试剂 3.1 盐酸 3.2 盐酸溶液HCl 3.3 乙醇95% 3?è?4g对二甲氨基苯甲醛溶于200 mL 95%乙醇和20mL 盐酸Array 中 155g/L剧毒)15.5g 溶于水中如叠氮化钠中含肼 将叠氮化钠溶于适量水中滤液置烧杯中不断搅拌待大部分叠氮化钠析出后经无水乙醇脱水后 2放在干燥器内冷却 注意精制操作应在通风柜内进行 100mg/L2HCl)或0.4060g硫酸肼(N2H4 H2SO4) 3.2?¨á?ò?è?1000mL容量瓶中 3.7 肼标准溶液吸取肼标准贮备液(3.6)10.0mLó?HCl 溶液(3.2)稀释至标线 4.1 分光光度计 4.2 具塞比色管 5 试样制备 5.1 采样与贮存样品均使用玻璃瓶 用盐酸固定可保存24h ·?±e?óè???±ê×?èüòo(3.7)0.5 2.00 6.00 10.00mL加入10mL对二甲氨基苯甲醛溶液 混匀用10cm光程的比色皿于458nm 波长处测定吸光度
含量为横坐标绘制校准曲线 检查水样pH值 将水样调至7左右 加蒸馏水稀释至25mL标线20min 后用10cm光程的比色皿于458nm 波长处测定吸光度 6.3 空白试验 取10mL蒸馏水代替水样 7 结果计算 试样中肼含量C(mg/L)按下式计算 mìg 分析试样体积 8 精密度和准确度 8个实验室对0.1000.800mg/L的标准溶液结果如下 0.9% 8.2 再现性 三个浓度的实验室间相对标准偏差分别为4.4%0.9% 9 参考文献 GB/T 15507-1995 è????ù?Do?óD?¢D?μ?1ìì???á£?úè¥?aê???3?μ?êyoáéy???ùoó?òà?D?è¥3y?ó?ê è???±e???ùóD??12′?ê± 3.2?ù?ó1% 的碘化钾0.4mL混匀再按操作步骤(6.2)进行 用20%氢氧化钠溶液调至水样成红色于通风柜中加HCl (3.2)至红色消失 3.1再按操作步骤(6.2)进行 水样的酸度调到1N后 A3 计算 如测定结果以水合肼计因1.56份N2H4
实验分光光度法测定铁 The following text is amended on 12 November 2020.
实验十四邻二氮菲分光光度法测定铁的含量 一、实验目的 1.学习吸光光度法测量波长的选择方法; 2.掌握邻二氮菲分光光度法测定铁的原理及方法; 3. 掌握分光光度计的使用方法。 二、实验原理 分光光度法是根据物质对光选择性吸收而进行分析的方法,分光光度法用于定量分析的理论基础是朗伯比尔定律,其数学表达式为:A=εb C 邻二氮菲(又称邻菲罗啉)是测定微量铁的较好试剂,在pH=2~9的条件下,二价铁离子与试剂生成极稳定的橙红色配合物。摩尔吸光系数ε=11000 L·mol-1·cm-1。在显色前,用盐酸羟胺把Fe3+还原为Fe2+。 2Fe3++2NH 2OHHCl→2Fe2++N 2 +4H++2H 2 O+2Cl- Fe2+ + Phen = Fe2+ - Phen (橘红色) 用邻二氮菲测定时,有很多元素干扰测定,须预先进行掩蔽或分离,如钴、镍、铜、铅与试剂形成有色配合物;钨、铂、镉、汞与试剂生成沉淀,还有些金属离子如锡、铅、铋则在邻二氮菲铁配合物形成的pH范围内发生水解;因此当这些离子共存时,应注意消除它们的干扰作用。 三、仪器与试剂 1.醋酸钠:l mol·L-1; 2.盐酸:6 mol·L-1; 3.盐酸羟胺:10%(用时配制); 4.邻二氮菲(%):邻二氮菲溶解在100mL1:1乙醇溶液中; 5.铁标准溶液。 (1)100μg·mL-1铁标准溶液:准确称取(NH 4) 2 Fe(SO 4 ) 2 ·12H 2 0于烧杯中, 加入20 mL 6 mol·L-1盐酸及少量水,移至1L容量瓶中,以水稀释至刻度,摇匀. 6.仪器:7200型分光光度计及l cm比色皿。 四、实验步骤 1.系列标准溶液配制 (1)用移液管吸取10mL100μg·mL-1铁标准溶液于100mL容量瓶中,加入2mL 6 mol·L-1盐酸溶液, 以水稀释至刻度,摇匀. 此溶液Fe3+浓度为10μg·mL-1. (2) 标准曲线的绘制: 取50 mL比色管6个,用吸量管分别加入0 mL,2 mL,4 mL, 6 mL, 8 mL和10 mL10μg·mL-l铁标准溶液,各加l mL盐酸羟胺,摇匀; 经再加2mL邻二氮菲溶液, 5 mL醋酸钠溶液,摇匀, 以水稀释至刻度,摇匀后放置 10min。 2.吸收曲线的绘制 取上述标准溶液中的一个, 在分光光度计上,用l cm比色皿,以水为参比溶液,用不同的波长,从440~560 nm,每隔10 nm测定一次吸光度,在最大吸收波长
【含量测定】照紫外-可见分光光度法(附录V A)测定。 1.仪器与测定条件:室温:____℃相对湿度:____% 分析天平编号:;水浴锅编号:; 紫外可见分光光度计编号:; 2.对照品溶液的制备: 取西贝母碱对照品适量,精密称定,加三氯甲烷制成每1ml含_______mg的溶液,即得。 3. 供试品溶液的制备: 取本品粉末(过三号筛)约______g,精密称定,置具塞锥形瓶中,加浓氨试液3ml,浸润1小时。加三氯甲烷-甲醇(4:1)混合溶液40ml,置80℃水浴加热回流2小时,放冷,滤过,滤液置50ml量瓶中,用适量三氯甲烷-甲醇(4:1)混合溶液洗涤药渣2~3次,洗液并入同一量瓶中,加三氯甲烷-甲醇(4:1)混合溶液至刻度,摇匀,即得。 4.标准曲线的制备: 精密量取对照品溶液0.1ml、0.2ml、0.4ml、0.6ml、1.0ml,置25ml具塞试管中,分别补加三氯甲烷至10.0ml,精密加水5ml、再精密加0.05%溴甲酚绿缓冲液(取溴甲酚绿0.05g,用0.2mol/L氢氧化钠溶液6ml使溶解,加磷酸二氢钾1g,加水使溶解并稀释至100ml,即得)2ml,密塞,剧烈振摇,转移至分液漏斗中,放置30分钟。取三氯甲烷液,用干燥滤纸滤过,取续滤液,以相应的试剂为空白。 5.测定法: 照紫外-可见分光光度法(附录ⅤA),在nm波长处测定吸光度,以吸光度为纵坐标,浓度为横坐标,绘制标准曲线。依法测定吸光度,从标准曲线上读出供试品溶液中含西贝母碱的重量,计算,即得。 6.结果与计算 6.1 标准曲线制备:
对照品批号 纯 度 S 对照品来源 干燥条件 对照品称重W 对(mg) 各浓度点稀释倍数f 对 溶液浓度C 对(ug/ml) 吸光度A 对 线性回归方程 A=( )C +/-( ) r =( ) 计算公式: W S C f ?= 对对对 C 对= 6.2 样品测定: 水分Q 取样量W 样(g ) 样品稀释倍数f 样 样品吸光度A 样 样品平均吸光度A 样 浓度C(ug/ml) 含量X (%) 平均含量X (%) 计算公式:() %100Q 110W f C X 6 ?-???= 样样 样 X 1= X 2= 7.本品按干燥品计算,含总生物碱以西贝母碱(C 27H 43NO 3)计,不得少于0.050%。 结果: 规定 检验人: 检验日期: 复核人: 复核日期:
离子色谱分析方法通则 离子色谱分析方法通则 1 范围 本标准规定了离子色谱法对仪器的要求和分析方法。所用仪器应具备输液泵、离子交换色谱柱、抑制器以及检测器(电导检测器、安培检测器、吸光度检测器或者其中任一种检测器)等。系统中应含完成分析任务所必需的附件—色谱工作站或积分仪等。 本标准适用于多种阴离子、阳离子、有机酸、糖类的测定。 2. 引用标准 GB 1.4-88 标准化工作导则化学分析方法标准编写规定 GB 3102.8-93 物理化学和分子物理学的量和单位 3 定义 3.1 电导conductance 电阻的倒数称为电导,单位为西门子,符号是S。它的导出单位为微西门子,符号是 口So 1S=106(i So 3.2 电导率conductivity 25°C时,一立方厘米液体的电阻的倒数,以Q 1 ? cm1或S/cm表示。 3.3 抑制电导检测suppressed conductance detection 在分离柱后,采用离子交换膜或离子交换柱将淋洗液中的淋洗离子转变为弱酸、弱碱或水,使淋洗液的背景电导降低,同时提高检测灵敏度的方法称为抑制电导检测。 3.4 分辨率(分离度) resolution 评价色谱柱对相邻双峰分离情况的指示:分峰的分离情 况。分辨率按 式中R —相邻两组分峰的分辨率 tR1 ——组分1 的保留时间 tR2 ——组分2 的保留时间
W1 ——组分1 的峰底宽度 W2 ——组分1 的峰底宽度 4 方法原理 不同的色谱柱中装填有不同类型的离子交换树脂。离子交换树脂上的活性交换基团能与样品中的离子及流动相中的淋洗离子发生离子交换作用。此种交换作用又因不同离子与树脂上的活性交换基团之间的静电力或亲和力存在差异,与树脂静电力或亲和力大的离子易被保留而难于被洗脱,静电力或亲和力小的离子则易于洗脱。随着淋洗液的流动,样品中的离子与树脂上的交换基团不断地发生交换—洗脱—再交换—再洗脱,最终被淋洗液带到检测器中形成高斯分布型色谱峰。在一定的色谱条件下组分峰的流出时间即保留时间固定,以此作为组分离子的定性依据。在一定的浓度范围内组分的峰面积(或峰高)正比于组分的浓度,积分仪拾得此信号给出组分的定量结果。 图 1 分辨率示意图 5 试剂和材料 5.1 配制淋洗液、再生液的试剂纯度应是分析纯(A.R)或分析纯以上试剂。 5.2 去离子水应满足以下要求: 5.2.1 电导率: 5.2.2 配制淋洗液前,去离子水应脱气5min 。 5.3 淋洗液、再生液、柱后衍生剂 5.3.1 淋洗液 5.3.1.1 1.8mmol/L , Na2CO3及1.7mmol/L NaHC03淋洗液:0.19g 无水Na2CO3和 0.14g NaHCO3溶于少量去离子水中,稀释至1000ml。用于阴离子分离。 5.3.1.2 15mmol/L Na2B4O7 : 7.6g 四硼酸钠(Na2B4O7- 10H2O溶解于去离子水中,稀释至1000ml。用于阴离子分离。 5.3.1.3 30mmol/L HCl :以去离子水稀释30ml 1mol/L HCl 于1000ml 容量瓶中。用于分离 Li+ 、Na+、NH4+、K+。 5.3.1.4 1mmol/L 乙二胺硝酸盐:60mg乙二胺(NH2CH2CH2NH溶于约950ml去离子水中,在酸度计上以3mol/L的硝酸调整该溶液的PH=4.8。最后定容到1000ml。用于分离Mg2+、Ca2+。 5.3.1.5 50mmol/L H2C2O4 及95mmol/L LiOH 淋洗液: 6.3g 乙二酸(草酸 H2C2O4 H2O), 4.0g氢氧化锂(LiOH - H2O溶解于去离子水中,稀释至1000ml。用于Cu2+、
本科毕业论文题目:亚甲基蓝分光光度法测定工业及 生活废水中的硫化物含量 学院:化学与化工学院 班级:06级化学五班 姓名:张翠云 指导教师:王海青职称:副教授完成日期:2010年06 月05 日
亚甲基蓝分光光度法测定工业及 生活废水中的硫化物含量 摘要:本文采用亚甲基蓝分光光度法测定工业及生活废水中硫化物的含量。实验结果表明:最低检出限浓度为0.02μg·mL-1,在0~25μg·mL-1范围内,相关系数r=0.9992,符合标准曲线对相关系数的要求(r>0.9990),即所测定硫化物含量具有真实性,是测定工业及生活废水中的硫化物含量的一种有效方法。 关键词:亚甲基蓝分光光度法;硫化物;水质分析
目录 1 引言 (1) 2 实验部分 (2) 2.1 实验原理 (2) 2.2 仪器和试剂 (2) 2.2.1 仪器 (2) 2.2.2 试剂 (3) 2.3 实验过程 (4) 2.3.1 水样采集及固定 (4) 2.3.2 标准曲线的绘制 (4) 2.3.3 样品的测定 (5) 2.3.4 空白测定 (6) 3 结果与讨论 (6) 3.1 实验结果分析 (6) 3.2 实验影响因素 (7) 3.2.1 水样预处理过程的影响 (7) 3.2.2 标定过程的影响 (7) 3.2.3 显色过程的影响 (8) 3.3 实验问题及解决 (8) 参考文献 (9) 致谢 (10)
1引言 此论文依据中华人民共和国环境保护行业标准GB-T 16489-1996,亚甲基蓝分光广度法测定水质硫化物[1,3]所写。该标准适用于地下水,化工,选矿等工业废水和生活废水中硫化物的测定,本次实验主要对大同地区矿水,甘河,高地,小站等四个采样点的水中硫化物进行测定。 我们通常说的水质硫化物系指水中溶解性的无机硫化物和酸溶性金属硫化物,具体包括溶解性的H2S、HS-、S2-以及存在悬浮物中的可溶性硫化物和酸可溶性金属硫化物和一些未电离的有机,无机类硫化物。它们是细菌在厌氧条件分解水中硫酸盐和有机含硫化合物而产生的。 由于这些硫化物随废水排出,往往以硫化氢的形式不断溢散于空气中,毒性很大。它可与人体细胞色素,氧化酶及该物质中的二硫键(-S-S)作用,影响细胞的氧化过程,造成细胞缺氧而危害人的生命,硫化氢除自身腐蚀金属外还可使污水中的生物氧化生成硫酸进而腐蚀下水道。因此硫化物的含量已成为水体污染的一项重要指标,近几年来水质硫化物的测定已成为一项常规的监测项目。 对于水和废水中硫化物的测定方法已有很多报道,属仪器测定方法的有荧光法,间接原子吸收法,紫外分光广度法,亚甲基蓝分光光度法等。属化学分析法的则有碘量法等各种容量滴定法。紫外分光广度法简便快速,但灵敏度较差;荧光法和间接原子吸收法有较好的选择性,主要用于废水中微量硫化物的测定,测定范围通常为0.007~0.8μg·mL-1。亚甲基蓝分光光度法和碘量法则是测定废水中硫化物的经典法。通常样品中硫化物含量小于1mg·L-1时采用前者,样品中硫化物含量大于1mg·L-1时采用后者。但由于某些废水成分复杂,干扰因素较多,用碘量法测定时有时存在较大误差,即碘量法的适用范围有一定限制,一般适用于硫化物含量较高且干扰因素较少的水样。而亚甲基蓝分光光度法由于灵敏度高,最低检出限浓度为0.02μg·mL-1,选择性好,加上予处理装置一般能消除干扰,因此作为首选方法。 采用该方法测定的关键几步:水样采集及固定;标准曲线的绘制;样品的测定;空白测定等。注意的是,由于水中硫化物含量与气象因素及环境因素等有关,即在硫化物的测定中,影响因素[12-14]很多,尤其是水样的采集固定以及
草甘膦常见制剂的区别 1.草甘膦品种 按存在方式分,有:草甘膦异丙胺盐,草甘膦酸铵盐,草甘膦酸钾盐,草甘膦酸钠盐,草甘膦二甲胺盐(新安开发,暂定)等; 2.按草甘膦离子的含量,41%异丙胺盐=30.5%草甘膦=3 3.5%草甘膦铵盐=37.5%草甘膦钾盐=3 4.5%草甘膦钠盐,二甲胺盐不详 3.在除草效果来说,钾盐稍大于异丙胺盐稍大于铵盐、钠盐。 4.草甘膦难溶于水,它的盐易溶于水,盐可以有钾盐、钠盐、铵盐、异丙胺盐等。草甘膦是草甘膦酸,属酸性,水溶性差,需要成盐,增加其水溶性,一般成铵盐和异丙胺盐,钾盐、钠盐等,最常见的是铵盐和异丙胺盐,41%的异丙胺盐,33%的铵盐中草甘膦含量都是30%,41%异丙胺盐需要用原药配置,而现在市面上标30%的草甘膦是不成盐的 企标中计算公式:草甘膦异丙胺盐=1.349*草甘膦含量,这个公式要先确定该盐是草甘膦异丙胺盐,而无其它形式的盐,这计算方法才勉强成立,但企标中没有这一点也就是说30%草甘膦水剂,不管是什么盐,都可以说成是41%草甘膦异丙胺盐水剂。 41%草甘膦异丙胺盐水剂实际上用不着制定企标,就用GB 20684-2006 草甘磷水剂。因为里面写得清清楚楚“本标准适用于由草甘膦原药或草甘膦可溶性盐和水及适宜的助剂组成的草甘膦水剂”。草甘膦的分子量为169.07,草甘膦异丙胺盐的分子量为228.2,
所以41%草甘膦异丙胺盐水剂实际上就是30.38%草甘膦水剂(略去小数点后面的数字就是30%草甘膦水剂)。有效成分是草甘膦。 41%的草甘膦异丙胺盐水剂和30%的草甘膦水剂还是有区别的: 41%草甘膦异丙胺盐水剂说明我的产品是异丙胺盐的 30%草甘膦水剂有以下几种可能: 1)30%草甘膦水剂以异丙胺盐的形式存在,类似于 40.5%草甘膦异丙胺盐水剂(略微底一点啊) 2)30%草甘膦水剂以钾盐形式存在,类似于37%的草甘膦钾盐水剂 3)30%草甘膦水剂以铵盐形式存在,类似于33%的草甘膦铵盐水剂 4)钠盐、二甲胺盐都有可能。 上述几个产品效果怎样,还要取决于使用助剂的种类,添加量。
紫外分光光度法测定未知物 1.仪器 1.1紫外分光光度计(UV-1801型);配石英比色皿(1cm)2个 1.2容量瓶(100mL):10个;容量瓶(250mL)1个 1.3吸量管(10mL、5mL):各1支 1.4移液管(20mL、25mL、50mL):各1支 2.试剂 2.1标准溶液(1mg/mL):维生素C、水杨酸、苯甲酸、山梨酸、邻二氮菲分别配成1mg/mL的标准溶液,作为储备液。 2.2未知液:浓度约为(40~60ug/mL)。(其必为给出的五种物质之一) 3.实验操作 3.1比色皿配套性检查 石英比色皿装蒸馏水,以一只比色皿为参比,在测定波长下调节透射比为100%,测定其余比色皿的透射比,其偏差应小于0.5%,可配成一套使用。 3.2未知物的定性分析 将五种标准储备液均稀释成10ug/mL的试液(配制方法由选手自定)。以蒸馏水为参比,于波长200~350nm范围内扫描五种溶液,绘制吸收曲线,根据所得到的吸收曲线对照标准谱图,确定被测物质的名称,并依据吸收曲线确定测定波长。五种标准物质溶液的吸收曲线参五种标准物质溶液的吸收曲线参五种标准物质溶液的吸收曲线参五种标准物质溶液的吸收曲线参考考考考附图附图附图附图。。。。 3.3未知物定量分析 根据未知液吸收曲线上测定波长处的吸光度,确定未知液的稀释倍数,并配制待测溶液3份,进行平行测定。 推荐方法 3.3.1维生素C含量的测定:准确吸取1mg/mL的维生素C标准储备液50.00mL,在250mL容量瓶中定容(此溶液的浓度为200ug/mL)。再分别准确移取1、2、4、6、8、10mL上述溶液,在100mL容量瓶中定容(浓度分别为2、4、8、12、16、20 ug/mL)。准确移取20.00mL维生素C未知液,在100mL容量瓶中定容,于
41%异丙胺盐与30%草甘膦的区别 10%的草甘膦马上就要退出市场了,这几天几乎所有的农药经销商都在找30%的草甘膦,今天下午跟一个市场“地头蛇”讨论草甘膦问题正欢,突然来了一个山东某农资企业的业务员向该客户推广他们企业的主打产品30%的草甘膦,客户看了看包装说:你们的包装不错嘛,价位也不错,我现在正好缺一个这样的产品去占市场。。。。不过,你们这个30%的草甘膦下面海标注了41%的草甘膦异丙胺盐,这个异丙胺盐跟草甘膦有什么区别?有什么好处?。。。只见那个业务员的脸色由白转红,又由红转惨白,结果嘴里哆嗦着,不知道说什么好了,我看他估计是不知道怎么说,或者是不懂,我就详细的向客户解释了两者的区别和作用,结果,这个这可客户当场决定给我们草甘膦产品打120吨的预付款,当然把那个山东的企业业务员拒之门外了。。。我暗自庆幸,幸好昨晚恶补了下除草剂知识啊。。。。我这里正好有一份资料,拿出来给大家分享下,我相信搞不清楚这几者之间的区别和联系的人,大有人在,我们共同进步吧! 按化学分类,草甘膦异丙胺盐、草甘膦铵盐、草甘膦分属植物源、季胺盐和有机磷类。 存在方式也不一样,41%异丙胺盐=30.5%草甘膦=33.5%草甘膦铵盐=37.5%草甘膦钾盐=34.5%草甘膦钠盐 在除草效果来说,钾盐稍大于异丙胺盐,异丙胺盐稍大于铵盐、钠盐。 草甘膦难溶于水,它的盐易溶于水,盐可以有钾盐、钠盐、铵盐、异丙胺盐等。草甘膦是草甘膦酸,属酸性,水溶性差,需要成盐,增加其水溶性,一般成铵盐和异丙胺盐,钾盐、钠盐等,最常见的是铵盐和异丙胺盐,41%的异丙胺盐,33%的铵盐中草甘膦含量都是30%,41%异丙胺盐需要用原药配置,而现在市面上标30%的草甘膦是不成盐的。 企标中计算公式:草甘膦异丙胺盐=1.349*草甘膦含量,这个公式要先确定该盐是草甘膦异丙胺盐,而无其它形式的盐,这计算方法才勉强成立,但企标中没有这一点。也就是说30%草甘膦水剂,不管是什么盐,都可以说成是41%草甘膦异丙胺盐水剂。 41%草甘膦异丙胺盐水剂实际上用不着制定企标,就用GB 20684-2006 草甘磷水剂。因为里面写得清清楚楚“本标准适用于由草甘膦原药或草甘膦可溶性盐和水及适宜的助剂组成的草甘膦水剂”。草甘膦的分子量为169.07,草甘膦异丙胺盐的分子量为228.2,所以41%草甘膦异丙胺盐水剂实际上就是30.38%草甘膦水剂(略去小数点后面的数字就是30%草甘膦水剂)。有效成分是草甘膦。 41%的草甘膦异丙胺盐水剂和30%的草甘膦水剂还是有区别的: 41%草甘膦异丙胺盐水剂说明我的产品是异丙胺盐的 30%草甘膦水剂有以下几种可能: 1)30%草甘膦水剂以异丙胺盐的形式存在,类似于40.5%草甘膦异丙胺盐水剂(略微底一点啊)2)30%草甘膦水剂以钾盐形式存在,类似于37%的草甘膦钾盐水剂 3)30%草甘膦水剂以铵盐形式存在,类似于33%的草甘膦铵盐水剂 4)钠盐、二甲胺盐都有可能。
紫外分光光度法测定蛋白质含量 一、实验目的 1.学习紫外光度法测定蛋白质含量的原理; 2.掌握紫外分光光度法测蛋白质含量的实验技术。 二、实验原理 1.测蛋白质含量的方法主要有:①测参数法:折射率、相对密度、紫外吸收等;②基于化学反应:定氮法、双缩脲法、Folin―酚试剂法等。本实验采用紫外分光光度法。 2.蛋白质中的酪氨酸和色氨酸残基的苯环中含有共轭双键,因此,蛋白质具有吸收紫外光的性质,其最大吸收峰位于280nm附近(不同蛋白质略有不同)。在最大吸收波长处,吸光度与蛋白质溶液的浓度服从朗伯―比尔定律。 利用紫外吸收法测蛋白质含量的准确度较差,原因有二:①对于测定那些与标准蛋白质中酪氨酸和色氨酸含量差异较大的蛋白质,有一定误差,故该法适于测定与标准蛋白质氨基酸组成相似的蛋白质;②样品中含有的嘌呤、嘧啶等吸收紫外光的物质,会出现较大干扰。 三、仪器与试剂 TU―1901紫外可见分光光度计、标准蛋白质溶液3.00mg·mL-1、0.9%NaCl 溶液、试样蛋白质溶液。 10mL比色管、1cm石英比色皿、吸量管。 四、实验步骤 1.绘制吸收曲线 用吸量管吸取2mL3.00mg·mL-1标准蛋白质溶液于10mL比色管中,用0.9%NaCl溶液稀释至刻度,摇匀。用1cm石英比色皿,以0.9%NaCl溶液作参比溶液,在190~400nm间每隔5nm测一次吸光度Abs,记录数据并作图。 2.绘制标准曲线 用吸量管分别吸取1.0、1.5、2.0、2.5、3.0mL3.00mg·mL-1标准蛋白质溶液于10mL比色管中,用0.9%NaCl溶液稀释至刻度,摇匀。用1cm石英比色皿,以0.9%NaCl溶液作参比溶液,在波长280nm处分别测其吸光度,记录数据并作图。 3.样品测定 取适量浓度试样蛋白质溶液,在波长280nm处测其吸光度,重复三次。在已经得到标准曲线的情况下,为了使测量结果准确度高,待测溶液的浓度需在标准曲线的线性范围内,所以,先测定试样蛋白质原液的吸光度(1.363),估算浓度为2.0960 mg·mL-1,再将原试液稀释至5倍(即取2mL试液,用0.9%NaCl 溶液稀释至刻度,摇匀),估算浓度为0.4192 mg·mL-1,测吸光度,重复三次五、数据处理与结果分析