
细菌16S rDNA序列比对进化树构建
生物科学131班 13213103王馨悦
一、实验目的
学习并掌握使用MEGA软件构建细菌16S rDNA的进化树
二、实验原理
1、细菌识别与鉴定手段的发展
1)传统的表型观察:群体(菌落)、个体
2)生理生化分类:如格兰仕阳性或阴性细菌的鉴定
3)分子水平鉴定:如(G+C)mol%、16SrDNA等,具有耗时短,成本低,
准确性高的优点
2、rRNA的特性
1)具有重要且恒定的生理功能;
2)约占细胞中RNA含量的90%,易于提取
3、细菌中包括三种rRNA,分别为5S rRNA、16S rRNA、23S rRNA
1)5S rRNA核苷酸太少,没有足够的遗传信息用于分类研究
2)23S rRNA核苷酸数几乎是16S rRNA的两倍,分析较困难
3)16S rRNA适于作为序列分析对象的依据
a.在16S rRNA分子中,既含有高度保守的序列区域,又有中度保守和高度
变化的序列区域,因而它适用于进化距离不同的各类生物亲缘关系的研究;
b.16S rRNA分子分子量大小适中,约1540bp,便于序列分析;
c. 16S rRNA普遍存在于真核生物和原核生物中(真核生物中其同源分子是18S rRNA)。因此它可以作为测量各类生物进化的工具。
三、实验步骤
1、NCBI序列下载
根据实验中老师提供的Genbank登录号(EF012357,AF506513,AB017203)在NCBI上 (https://www.doczj.com/doc/916727035.html,/blast/Blast.cgi)下载菌株的序列
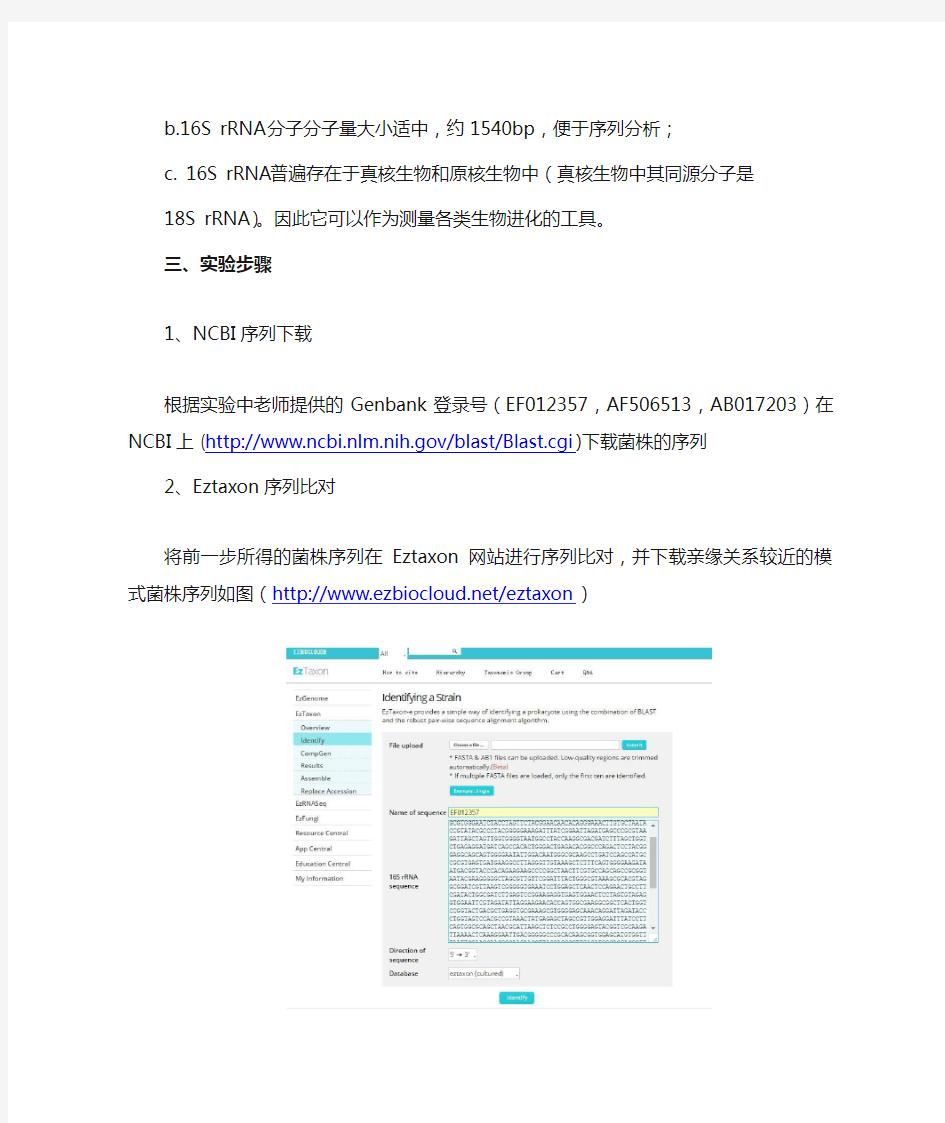
2、Eztaxon序列比对
将前一步所得的菌株序列在Eztaxon网站进行序列比对,并下载亲缘关系
较近的模式菌株序列如图(https://www.doczj.com/doc/916727035.html,/eztaxon)
a.选择同属亲缘关系较近的模式菌株
选择的原则:依据同源关系由近而远,代表每个种的菌株一到三个左右,尽可能选择同属的菌株,同属菌株很少的,可以选择近缘属的模式菌株或其他代表菌株
b.将所选序列(Fasta格式文件)下载保存
3、MEGA构建进化树
四、实验结果
1、EF012357
五、总结与收获
通过本次实验
我对16S rRNA用于进化分析的原理及意义有了全面深刻的了解和体会;并且在多次练习中初步掌握了MEGA软件结合NCBI数据库和Eztaxon模式菌株网站进行的序列查找、选择、比对、构树及修饰等操作;学会了构建进化树反映生物间的亲缘关系。此外,我在本次实验中对生物信息学在微生物学科的重要应用价值有了全新的认识,激励自己进行多分支学科知识的扩充和运用。
用16S-rDNA方法鉴定细菌种属
用16S rDNA方法鉴定细菌种属 一、实验目的 1. 掌握16S rDNA对细菌进行分类的原理及方法; 2. 掌握DNA提取、PCR原理及方法、DNA片段回收等实验操作。 二、实验原理 细菌rRNA(核糖体RNA)按沉降系数分为3种,分别为5S、16S和23S rRNA。16S rDNA是细菌染色体上编码16S rRNA相对应的DNA序列,存在于所有细菌染色体基因中。 16SrDNA鉴定是指用利用细菌16SrDNA序列测序的方法对细菌进行种属鉴定。包括细菌基因组DNA提取、16SrDNA特异引物PCR扩增、扩增产物纯化、DNA 测序、序列比对等步骤。是一种快速获得细菌种属信息的方法。 16S rDNA是细菌的系统分类研究中最有用的和最常用的分子钟,其种类少,含量大(约占细菌RNA 含量的80%),分子大小适中,存在于所有的生物中,其进化具有良好的时钟性质,在结构与功能上具有高度的保守性,素有“细菌化石”之称。在大多数原核生物中rDNA都具有多个拷贝,5S、16S、23S rDNA的拷贝数相同。16S rDNA由于大小适中,约1.5Kb左右,既能体现不同菌属之间的差异,又能利用测序技术较容易地得到其序列,故被细菌学家和分类学家接受。
16SrRNA 的编码基因是16SrDNA ,但是要直接将 16SrRNA 提取出来很困难,因为易被广泛存在的RNase 降解,因而利用16S rDNA 鉴定细菌,其技术路线如下: 细菌基因组的提取: PCR 的基本原理 : PCR 技术的基本原理 类似于DNA 的天然复制过程,其特异性依赖于与靶序列两端互补的 寡核苷酸引物。PCR 由变性--退火--延伸三个基本反应步骤构成: ①模板DNA 的变性:模板DNA 经加热至93℃左右一定时间后,使模板DNA 双链或经PCR 扩增形成的双链DNA 解离,使之成为单链,以便它与引物结合,为下轮反应作准备; ②模板DNA 与引物的退火(复性):模板DNA 经加热变性成单链后,温度降至55℃左右,引 物与模板DNA 单链的互补序列配对结合; 纯化DNA 分离出DNA
【转载】分子进化树构建及数据分析的简介 分子进化树构建及数据分析的简介 mediocrebeing, rodger, lylover[1], klaus, oldfish, yzwpf [1] lylover. Email: lylover_2005@https://www.doczj.com/doc/916727035.html, 一、引言 开始动笔写这篇短文之前,我问自己,为什么要写这样的文章?写这样的文章有实际的意义吗?我希望能够解决什么样的问题?带着这样的疑惑,我随手在丁香园(DXY)上以关键字“进化分析求助”进行了搜索,居然有289篇相关的帖子(2006年9月12日)。而以关键字“进化分析”和“进化”为关键字搜索,分别找到2,733和7,724篇相关的帖子。考虑到有些帖子的内容与分子进化无关,这里我保守的估计,大约有3,000~4,000篇帖子的内容,是关于分子进化的。粗略地归纳一下,我大致将提出的问题分为下述的几类:1.涉及基本概念。例如,“分子进化与生物进化是不是一个概念”,“关于微卫星进化模型有没有什么新的进展”以及“关于Kruglyak的模型有没有改进的出现”,等等。 2.关于构建进化树的方法的选择。例如,“用boostrap NJ得到XX图,请问该怎样理解?能否应用于文章?用boostrap test中的ME法得到的是XXX树,请问与上个树比,哪个更好”,等等。 3.关于软件的选择。例如,“想做一个进化树,不知道什么软件能更好的使用且可以说明问题,并且有没有说明如何做”,“拿到了16sr RNA数据,打算做一个系统进化树分析,可是原来没有做过这方面的工作啊,都要什么软件”,“请问各位高手用clustalx做出来的进化树与phylip做的有什么区别”,“请问有做过进化树分析的朋友,能不能提供一下,做树的时候参数的设置,以及代表的意思。还有各个分支等数值的意思,说明的问题等”,等等。 4.蛋白家族的分类问题。例如,“搜集所有的关于一个特定domain的序列,共141条,做的进化树不知具体怎么分析”,等等。 5.新基因功能的推断。例如,“根据一个新基因A氨基酸序列构建的系统发生树,这个进化树能否说明这个新基因A和B同源,属于同一基因家族”,等等。 6.计算基因分化的年代。例如,“想在基因组水平比较两个或三个比较接近物种之间的进化年代的远近,具体推算出他们之间的分歧时间”,“如何估计病毒进化中变异所需时间”,等等。 7.进化树的编辑。例如生成的进化树图片,如何进行后续的编辑,比如希望在图片上标注某些特定的内容,等等。 由于相关的帖子太多,作者在这里对无法阅读全部的相关内容而致以歉意。同时,作者归纳的这七个问题也并不完全代表所有的提问。对于问题1所涉及到的基本的概念,作者推荐读者可参考由Masatoshi Nei与Sudhir Kumar所撰写的《分子进化与系统发育》(Molecular Evolution and Phylogenetics)一书,以及相关的分子进化方面的最新文献。对于问题7,作者之一lylover一般使用Powerpoint 进行编辑,而Photoshop、Illustrator及Windows自带的画图工具等都可以使用。
构建系统发育树需要注意的几个问题 1 相似与同源的区别:只有当序列是从一个祖先进化分歧而来时,它们才是同源的。 2 序列和片段可能会彼此相似,但是有些相似却不是因为进化关系或者生物学功能相近的缘故,序列组成特异或者含有片段重复也许是最明显的例子;再就是非特异性序列相似。 3 系统发育树法:物种间的相似性和差异性可以被用来推断进化关系。 4 自然界中的分类系统是武断的,也就是说,没有一个标准的差异衡量方法来定义种、属、科或者目。 5 枝长可以用来表示类间的真实进化距离。 6 重要的是理解系统发育分析中的计算能力的限制。任何构树的实验目的基本上就是从许多不正确的树中挑选正确的树。 7 没有一种方法能够保证一颗系统发育树一定代表了真实进化途径。然而,有些方法可以检测系统发育树检测的可靠性。第一,如果用不同方法构建树能得到同样的结果,这可以很好的证明该树是可信的;第二,数据可以被重新取样(bootstrap),来检测他们统计上的重要性。 分子进化研究的基本方法 对于进化研究,主要通过构建系统发育过程有助于通过物种间隐含的种系关系揭示进化动力的实质。 表型的(phenetic)和遗传的(cladistic)数据有着明显差异。Sneath和Sokal(1973)将表型性关系定义为根据物体一组表型性状所获得的相似性,而遗传性关系含有祖先的信息,因而可用于研究进化的途径。这两种关系可用于系统进化树(phylogenetictree)或树状图(dendrogram)来表示。表型分枝图(phenogram)和进化分枝图(cladogram)两个术语已用于表示分别根据表型性的和遗传性的关系所建立的关系树。进化分枝图可以显示事件或类群间的进化时间,而表型分枝图则不需要时间概念。文献中,更多地是使用“系统进化树”一词来表示进化的途径,另外还有系统发育树、物种树(species tree)、基因树等等一些相同或含义略有差异的名称。 系统进化树分有根(rooted)和无根(unrooted)树。有根树反映了树上物种或基
16S rDNA鉴定细菌的方法 细菌16S rDNA鉴定主要分为7个部分: 1.提取细菌基因组DNA, 2.设计/选择引物进行PCR扩增,电泳检测纯度与大小。 3.琼脂糖凝胶电泳分离 4.胶回收目的片段 5.目的片段测序。 6.BLAST比对获取相似片段。 7.构建系统进化树 试剂: 1.1培养基:通常选择组分简单且细菌生长良好的培养基(培养基组分过于复杂会影响DNA 的提取效果,也可以在裂解细菌前用TE缓冲液对菌体进行洗涤。)。 1.2 1M Tris-HCl (pH7.4, 7.6, 8.0)(1L):121.1g Tris,加浓盐酸约(70ml, 60ml, 42ml),高温高压灭菌后,室温保存。冷却到室温后调pH,每升高1℃,pH大约下降0.03个单位。 1.3 0.5M EDTA(pH8.0)(1L):186.1g Na2EDTA?2H2O,用NaOH调pH至8.0(约20g),高温高压灭菌,室温保存。 1.410×TE Buffer(pH7.4,7.6,8.0)(1L):组分:100 mM Tris-HCl,10 mM EDTA。1M Tris-HCl (pH7.4,7.6,8.0)取100ml,0.5M EDTA(pH8.0)取20ml。高温高压灭菌,室温保存。1×TE Buffer用10×TE Buffer稀释10倍即可。 1.5 10%SDS(W/V):称10g,68℃加热溶解,用浓盐酸调pH至7.2。室温保存。用之前在65℃溶解。配置时要戴口罩。 6、5M NaCl:称292.2gNaCl,高温高压灭菌,4℃保存。 7、CTAB/NaCl(10%CTAB,0.7M NaCl):溶解4.1g NaCl,加10g CTAB(十六烷基三甲基溴化铵),加热搅拌。用之前在65℃溶解。 8、氯仿/异戊醇:按氯仿:异戊醇=24:1(V/V)的比例加入异戊醇。 9、酚/氯仿/异戊醇(25:24:1):按苯酚与氯仿/异戊醇=1:1的比例混合Tris-HCl平衡苯酚与氯仿/异戊醇。 10、TAE缓冲液:使用液1×:0.04 mol/L Tris-乙酸,0.001 mol/L EDTA。浓储存液50×:242g Tris,57.1 ml 冰醋酸,100 ml 0.5 mol/L EDTA (pH8.0)。 11、6×上样缓冲液(100 ml):0.25%溴酚蓝(BPB),40%蔗糖,10 mmol/L EDTA (pH8.0)(0.2 ml),4℃保存。 12、0.6%琼脂糖凝胶:称取0.3g琼脂糖用TAE溶液配置50 ml。 13、EB:10 mg/ml。称取1g溴化乙锭定容至100ml。棕色瓶室温避光保存。EB的工作浓度为0.5ug/ml。当配置50ml 琼脂糖凝胶时加入EB为2.5ul。(因EB是剧毒物质,目前很多实验室用生物荧光染料替代,常用的有Gelred等) 14、蛋白酶K:20 mg/ml 溶于水,-20℃保存,反应浓度50 ug/ml,反应缓冲液:0.01 mol/L Tris (pH 7.8), 0.005 mol/L EDTA, 5% SDS,反应温度37-56℃。无需预处理。 15、RNase A:10 mg/ml。25 mg RNase A 加1M Tris(pH 7.5)25ul,2.5M NaCl 15ul,无菌水2460 ul,于100℃加热15分钟,缓慢冷却至室温,分装成小份保存于-20℃。(为避
一、实验原理 随着分子生物学的迅速发展,细菌的分类鉴定从传统的表型、生理生化分类进入到各种基因型分类水平,如(G+C)mol%、DNA杂交、rDNA指纹图、质粒图谱和16S rDNA序列分析等。 细菌中包括有三种核糖体RNA,分别为5S rRNA、16S rRNA、23S rRNA,rRNA基因由保守区和可变区组成。16S rRNA对应于基因组DNA上的一段基因序列称为16S rDNA。5S rRNA虽易分析,但核苷酸太少,仅几十bp,没有足够的遗传信息用于分类研究;23S rRNA含有的核苷酸数几乎是16S rRNA的两倍,分子量太大,分析较困难。而16S rRNA相对分子量在2kb左右,较为适合PCR 扩增,又具有保守性和存在的普遍性等特点,序列变化与进化距离相适应,序列分析的重现性极高,因此,现在一般普遍采用16S rRNA作为序列分析对象对微生物进行测序分析。 16SrRNA的编码基因是16SrDNA,但是要直接将16SrRNA提取出来很困难,因为易被广泛存在的RNase降解,因而利用16S rDNA鉴定细菌,其技术路线如下: PCR实验原理
即聚合酶链式反应,是指在DNA聚合酶催化下,以母链DNA为模板,以特定引物为延伸起点,通过变性、退火、延伸等步骤,体外复制出与母链模板DNA互补的子链DNA的过程。是一项DNA体外合成放大技术,能快速特异地在体外扩增任何目的DNA。 二、主要器具及试剂 PCR、电泳系统、DNA提取体系、Taq Polymerase、DNA Marker,溶菌酶、dNTP和E.coli JM109感受态细胞、pMD18-T Vector、琼脂糖、SDS裂解缓冲液、50×TAE电泳缓冲液贮存液、1×TE(pH 8.0) 三、操作方法 1. 细菌基因组总DNA的提取 接种纯化的菌株于LB液体培养基中,180 r/min,37 ℃培养过夜,按以下的方法提取细菌基因组总DNA。 (1)菌体收集:取1.5 mL新鲜的菌液于EP管中,12000 r/min离心30 s,弃净上清,收集菌体。 (2)辅助裂解:加100 μg/mL溶菌酶50 μL,37 ℃处理30min。 (3)裂解:向每管加入200 mL预冷的SDS裂解缓冲液,用吸管头迅速强烈抽吸以悬浮和裂解细菌细胞。 (4)向每管加入66 μL 5 mol/L NaCl,充分混匀后,12000 r/min离心10 min,除去蛋白质复合物及细胞壁等残渣。 (5)将上清转移到新EP管中,加入等体积的Tris-饱和酚,充分混匀,12000 r/min离心3 min,进一步沉淀蛋白质。 (6)取离心后的水层,加等体积的氯仿/异戊醇(体积比24:1),充分混匀后,12000 r/min离心3 min,去除苯酚。 (7)小心取上清,用预冷2倍体积的无水乙醇沉淀DNA,13000 r/min离心15 min,弃上清。 (8)用400 μL75%的乙醇洗涤沉淀2次。 (9)室温干燥后,用40 μL 1×TE溶解DNA。 (10)1.0%琼脂糖凝胶电泳检测基因组DNA。 (11)提取的基因组总DNA-40 ℃冰箱保存备用。
大家好: 我在此介绍几个进化树分析及其相关软件的使用和应用范围。这几个软件分别是PHYLIP、PUZZLE、PAUP、TREEVIEW、CLUSTALX和PHYLO-WIN (LINUX)。 在介绍软件之前,我先简要地叙述一下有关进化树分析的一些方法学问题。进化树也称种系树,英文名叫“Phyligenetic tree”。对于一个完整的进化树分析需要以下几个步骤:⑴要对所分析的多序列目标进行排列(To align sequences)。做ALIGNMENT的软件很多,最经常使用的有CLUSTALX和CLUSTALW,前者是在WINDOW下的而后者是在DOS下的。⑵要构建一个进化树(To reconstrut phyligenetic tree)。构建进化树的算法主要分为两类:独立元素法(discrete character methods)和距离依靠法(distance methods)。所谓独立元素法是指进化树的拓扑形状是由序列上的每个碱基/氨基酸的状态决定的(例如:一个序列上可能包含很多的酶切位点,而每个酶切位点的存在与否是由几个碱基的状态决定的,也就是说一个序列碱基的状态决定着它的酶切位点状态,当多个序列进行进化树分析时,进化树的拓扑形状也就由这些碱基的状态决定了)。而距离依靠法是指进化树的拓扑形状由两两序列的进化距离决定的。进化树枝条的长度代表着进化距离。独立元素法包括最大简约性法(Maximum Parsimony methods)和最大可能性法(Maximum Likelihood methods);距离依靠法包括除权配对法(UPGMAM)和邻位相连法(Neighbor-joining)。⑶对进化树进行评估。主要采用Bootstraping法。进化树的构建是一个统计学问题。我们所构建出来的进化树只是对真实的进化关系的评估或者模拟。如果我们采用了一个适当的方法,那么所构建的进化树就会接近真实的“进化树”。模拟的进化树需要一种数学方法来对其进行评估。不同的算法有不同的适用目标。一般来说,最大简约性法适用于符合以下条件的多序列:i 所要比较的序列的碱基差别小,ii 对于序列上的每一个碱基有近似相等的变异率,iii 没有过多的颠换/转换的倾向,iv 所检验的序列的碱基数目较多(大于几千个碱基);用最大可能性法分析序列则不需以上的诸多条件,但是此种方法计算极其耗时。如果分析的序列较多,有可能要花上几天的时间才能计算完毕。UPGMAM(Unweighted pair group method with arithmetic mean)假设在进化过程中所有核苷酸/氨基酸都有相同的变异率,也就
用16S rDNA方法鉴定细菌种属 一、实验目的 1. 掌握16S rDNA对细菌进行分类的原理及方法; 2. 掌握DNA提取、PCR原理及方法、DNA片段回收等实验操作。 二、实验原理 随着分子生物学的迅速发展,细菌的分类鉴定从传统的表型、生理生化分类进入到各种基因型分类水平,如(G+C)mol%、DNA杂交、rDNA指纹图、质粒图谱和16S rDNA序列分析等。 细菌中包括有三种核糖体RNA,分别为5S rRNA、16S rRNA、23S rRNA,rRNA基因由保守区和可变区组成。16S rRNA对应于基因组DNA上的一段基因序列称为16S rDNA。5S rRNA虽易分析,但核苷酸太少,没有足够的遗传信息用于分类研究;23S rRNA含有的核苷酸数几乎是16S rRNA的两倍,分析较困难。而16S rRNA相对分子量适中,又具有保守性和存在的普遍性等特点,序列变化与进化距离相适应,序列分析的重现性极高,因此,现在一般普遍采用16S rRNA作为序列分析对象对微生物进行测序分析。 利用16S rDNA鉴定细菌的技术路线: PCR的基本原理:必须已知部分序列设计出引物,10-30bp。
以此类推,呈几何级增长。一般进行25-35个扩增循环 DNA可扩增106~109倍。 三、实验步骤 (一)细菌基因组DNA提取 技术路线: 具体步骤: 1. 挑单菌落接种到10 mL LB培养基中37℃振荡过夜培养。(前期由老师完成) 2. 取2 mL培养液到2 mL Eppendorf管中,8000 rpm离心2分钟后倒掉上清液。 3. 再往同一离心管中2 mL培养液到2 mL Eppendorf管中,8000 rpm离心2分钟后倒掉上清液。 4. 加入200 μL 缓冲液GA,充分悬浮、混匀,将菌体彻底悬浮。 5. 再加入20 μL Protein K,混匀,55℃温育10分钟。(55℃为酶最适温度) 6. 加入220 μL缓冲液GB,混匀后,55℃温育10分钟。(实为50-60℃之间) 7. 加入220 μL 无水乙醇(降低体系粘稠度),充分混匀。将上清(挑去其中絮状杂质,少部分,会产生气泡,除不去))小心转入UNIQ-10柱(羟基纤维素膜,特异吸附核酸,因为吸附有限,所以损失核酸)中。13000 rpm(离心机最高设定实为10000rpm)离心5分钟,倒弃收集管内的液体。 6. 加入500 μL GD Solution,10000 rpm离心1分钟。
1.准备序列文件 准备fasta格式序列文件(fasta格式:大于号>后紧跟序列名,换行后是序列。举例如下)。每条序列可以单独为一个文件,也可以把所有序列放在同一文件内。 核酸序列: >sequence1_name CCTGGCTCAGGATGAACGCT 氨基酸序列: >sequence2_name MQSPINSFKKALAEGRTQIGF 2.多序列比对 打开MEGA 5,点击Align,选择Edit/Build Alignment,选择Create a new alignment,点击OK。
这时需要选择序列类型,核酸(DNA)或氨基酸(Protein)。 选择之后,在弹出的窗口中直接Ctrl + V粘贴序列(如果所有序列在同一个文件中,即可全选序列,复制)。也可以:点击Edit,选择Insert Sequence From File,选择序列文件(可多选)。
序列文件加载之后,呈蓝色背景(为选中状态)。点击按钮,选择Align DNA (如果是氨基酸序列,则会出现Align Protein)。弹出的窗口中设置比对参数,一般都是采用默认参数即可。点击OK,开始多序列比对。
比对完成后,呈现以下状态。 这时需要截齐两端含有---的序列:选中含有---的序列,按键Delete删除(注意:两端都需要截齐)。截齐之后,保存文件为:filename.mas
3.构建系统进化树 多序列比对窗口,点击Data,选择Phylogenetic Analysis,弹出窗口询问:所用序列是否编码蛋白质,根据实际情况选择Yes或No。此时,多序列比对文件就激活了,可以返回MEGA 5主界面建树了。
一、实验原理 随着分子生物学的迅速发展,细菌的分类鉴定从传统的表型、生理生化分类进入到各种基因型分类水平,如(G+C)mol%、DNA杂交、rDNA指纹图、质粒图谱与16S rDNA序列分析等。 细菌中包括有三种核糖体RNA,分别为5S rRNA、16S rRNA、23S rRNA,rRNA 基因由保守区与可变区组成。16S rRNA对应于基因组DNA上的一段基因序列称为16S rDNA。5S rRNA虽易分析,但核苷酸太少,仅几十bp,没有足够的遗传信息用于分类研究;23S rRNA含有的核苷酸数几乎就是16S rRNA的两倍,分子量太大,分析较困难。而16S rRNA相对分子量在2kb左右,较为适合PCR扩增,又具有保守性与存在的普遍性等特点,序列变化与进化距离相适应,序列分析的重现性极高,因此,现在一般普遍采用16S rRNA作为序列分析对象对微生物进行测序分析。 16SrRNA的编码基因就是16SrDNA,但就是要直接将16SrRNA提取出来很困难,因为易被广泛存在的RNase降解,因而利用16S rDNA鉴定细菌,其技术路线如下: PCR实验原理 即聚合酶链式反应,就是指在DNA聚合酶催化下,以母链DNA为模板,以特定
引物为延伸起点,通过变性、退火、延伸等步骤,体外复制出与母链模板DNA互补的子链DNA的过程。就是一项DNA体外合成放大技术,能快速特异地在体外扩增任何目的DNA。 二、主要器具及试剂 PCR、电泳系统、DNA提取体系、Taq Polymerase、DNA Marker,溶菌酶、dNTP与E、coli JM109感受态细胞、pMD18-T Vector、琼脂糖、SDS裂解缓冲液、50×TAE电泳缓冲液贮存液、1×TE(pH 8、0) 三、操作方法 1、细菌基因组总DNA的提取 接种纯化的菌株于LB液体培养基中,180 r/min,37 ℃培养过夜,按以下的方法提取细菌基因组总DNA。 (1)菌体收集:取1、5 mL新鲜的菌液于EP管中,12000 r/min离心30 s,弃净上清,收集菌体。 (2)辅助裂解:加100 μg/mL溶菌酶50 μL,37 ℃处理30min。 (3)裂解:向每管加入200 mL预冷的SDS裂解缓冲液,用吸管头迅速强烈抽吸以悬浮与裂解细菌细胞。 (4)向每管加入66 μL 5 mol/L NaCl,充分混匀后,12000 r/min离心10 min,除去蛋白质复合物及细胞壁等残渣。 (5)将上清转移到新EP管中,加入等体积的Tris-饱与酚,充分混匀,12000 r/min 离心3 min,进一步沉淀蛋白质。 (6)取离心后的水层,加等体积的氯仿/异戊醇(体积比24:1),充分混匀后,12000 r/min离心3 min,去除苯酚。 (7)小心取上清,用预冷2倍体积的无水乙醇沉淀DNA,13000 r/min离心15 min,弃上清。 (8)用400 μL75%的乙醇洗涤沉淀2次。 (9)室温干燥后,用40 μL 1×TE溶解DNA。 (10)1、0%琼脂糖凝胶电泳检测基因组DNA。 (11)提取的基因组总DNA-40 ℃冰箱保存备用。 2、PCR扩增细菌的16S rDNA
16SrDNA序列在微生物的分类鉴定上的应用 摘要 随着生物技术的迅速发展,16SrDNA分析技术在微生物分类鉴定及分子检测中得到广泛应用。根据16SrDNA的保守域和高变域,可以进行种属的鉴定。16SrDNA分析技术克服了传统生物培养法的限制,操作方便,检测快,准确度高且灵敏度高,已被广泛应用到菌种鉴定、群落对比分析、群落中系统发育及种群多样性的评估,是一种客观且可信度高的分类方法。 关键词 16SrDNA 微生物分类鉴定 传统的微生物分类是根据菌落的形态特征和生理生化特性,对菌种进行纯培养分离,然后从形态学、生理生化反应特征及免疫学特性加以鉴定。但近几十年来,传统微生物分类鉴定得到了巨大的革新,许多新技术和方法在微生物分类中得到广泛应用,使微生物分类鉴定从一般表型特征的鉴定,深化到遗传特性的鉴定。 1.微生物形态比较分类法的局限性 1.1 不能正确反映微生物形态 由于微生物菌群在进行纯培养时,不可避免地会造成菌株的富集或衰减,人为地改变了原始菌群的微生物生态构成,对研究结果造成了较大的偏差①。这样在应用上就只能局限于对简单环境下的微生物的研究,而对复杂环境下的微生物不能加以有效分析,严重影响了微生物基因组的研究。 1.2微生物资源大量丢失 许多研究研究表明,在自然环境中有相当多的菌种(约90%~99%)用纯培养方法无法培养出来。同时,纯培养分离方法采用配制简单的营养基质和固定的培养温度,忽略了气候变化和生物相互作用的影响,这种人工环境与原生境的偏差使得可培养的种类大大减少,仅占环境微生物总数的0.1%~1%②。因此,由于绝大多数微生物无法培养得到,丢失了大量微生物资源,对微生物多样性的认识较片面。 1.3不能保证分类的准确性和科学性 对形态特征、理化特征、菌体某些化学成分等特征完全相同时,可较准确的分类,但是对某特性中有某些差异的,往往难以判断。而且,方法繁琐耗时。 2. 16SrDNA及其鉴定依据 2.1 16SrDNA简介 16SrDNA是编码原核生物核糖体小亚基rRNA(16SrRNA)的基因,长度约1500bp,是细菌分类学研究中最常用、最有用的“分子钟”, 其序列包含10 个可变区( variable region ) 和与之相间的11 个恒定区( constantregion) ,可变区因细菌而异,且变异程度与细菌的系统发育密切相关。。Woese等③与Olsen等④基于16SrDNA的分析构建了现已被公认的全生命系统进化树。越来越多的细菌依据16SrDNA 被正确分类或重分类,尤其是许多环境中的细菌⑤、⑥ ,如Funke等将棒杆菌依赖细胞毒性1组及类似棒杆菌重新确定为放线菌的一个新种,即钮氏放线菌种, Bennasar等通过比较14株施氏假单胞菌的7个基因型(DNA2DNA杂交同源
1.MEGA构建系统进化树的步骤 2.CLUSTALX进行序列比对 1.MEGA构建系统进化树的步骤 1. 将要用于构建系统进化树的所有序列合并到同一个fasta格式文件,注意:所有序列的方向都要保持一致( 5’-3’)。如图: 2. 打开MEGA软件,选择"Alignment" - "Alignment Explorer/CLUSTAL",在对话框中选择Retrieve sequences from a file, 然后点OK,找到准备好的序列文件并打开,如图: 。 3. 在打开的窗口中选择”Alignment”-“Align by ClustalX” 进行对齐,对齐过程需要一段时间,对齐完成后,最好将序列两端切齐,选择两端不齐的部分,
单击右键,选择delete即可,如图: 。 4. 关闭当前窗口,关闭的时候会提示两次否保存,第一次无所谓,保存不保存都可以,第二次一定要保存,保存的文件格式是.meg。根据提示输入Title,然后会出现一个对话框询问是否是Protein-coding nucleotide sequence data, 根据情况选择Yes或No。最后出现一个对话框询问是否打开,选择Yes,如图: 。 5. 回到MEGA主窗口,在菜单栏中选择”Phylogeny”-“Bootstrap Test of Phylogeny” -“Neighbor-joining”,打开一个窗口,里面有很多参数可以设
置,如何设置这些参数请参考详细的MEGA说明书,不会设置就暂且使用默认值,不要修改,点击下面的Compute按钮,系统进化树就画出来了,如图: 在菜单栏中选择”Phylogeny”-“Bootstrap Test of Phylogeny” –“Minimun-evolution”,如图: 在菜单栏中选择”Phylogeny”-“Bootstrap Test of Phylogeny” –“Maximun-parsimony”,如图: 在菜单栏中选择”Phylogeny”-“Bootstrap Test of Phylogeny” –“UPGMA”,
多重序列比对及系统发生树的构建 作者:佚名来源:生物秀时间:2007-12-31 【实验目的】 1、熟悉构建分子系统发生树的基本过程,获得使用不同建树方法、建树材料和建树参数对建树结果影响的正确认识; 2、掌握使用Clustalx进行序列多重比对的操作方法; 3、掌握使用Phylip软件构建系统发生树的操作方法。 【实验原理】 在现代分子进化研究中,根据现有生物基因或物种多样性来重建生物的进化史是一个非常重要的问题。一个可靠的系统发生的推断,将揭示出有关生物进化过程的顺序,有助于我们了解生物进化的历史和进化机制。 对于一个完整的进化树分析需要以下几个步骤:⑴要对所分析的多序列目标进行比对(alignment)。⑵要构建一个进化树(phyligenetic tree)。构建进化树的算法主要分为两类:独立元素法(discrete character methods)和距离依靠法(distance methods)。所谓独立元素法是指进化树的拓扑形状是由序列上的每个碱基/氨基酸的状态决定的(例如:一个序列上可能包含很多的酶切位点,而每个酶切位点的存在与否是由几个碱基的状态决定的,也就是说一个序列碱基的状态决定着它的酶切位点状态,当多个序列进行进化树分析时,进化树的拓扑形状也就由这些碱基的状态决定了)。而距离依靠法是指进化树的拓扑形状由两两序列的进化距离决定的。进化树枝条的长度代表着进化距离。独立元素法包括最大简约性法(Maximum Parsimony methods)和最大可能性法(Maximum Likelihood methods);距离依靠法包括除权配对法(UPGMAM)和邻位相连法(Neighbor-joining)。⑶对进化树进行评估,主要采用Bootstraping法。进化树的构建是一个统计学问题,我们所构建出来的进化树只是对真实的进化关系的评估或者模拟。如果我们采用了一个适当的方法,那么所构建的进化树就会接近真实的“进化树”。模拟的进化树需要一种数学方法来对其进行评估。不同的算法有不同的适用目标。一般来说,最大简约性法适用于符合以下条件的多序列:i 所要比较的序列的碱基差别小,ii 对于序列上的每一个碱基有近似相等的变异率,iii 没有过多的颠换/转换的倾向,iv 所检验的序列的碱基数目较多(大于几千个碱基);
分子进化与系统进化树的构建 分子进化与系统进化树的构建 分子进化与系统进化树的构建 主要内容: 1、分子进化的研究方法 2、系统进化树的构建方法 3、系统进化树构建常用软件汇集 4、系统进化树构建方法及软件的选择 5、Phylip分子进化分析软件包简介及使用 6、如何利用MEGA3.1构建进化树 声明: 1、本篇涉及的资源主要源于网络及相关书籍,由酷友搜集、分析、整理、审改,供大家学习参考用,如有转载、传播请注明源于基因酷及本篇的工作人员;若本篇侵犯了您的版权或有任何不妥,请Email genecool@https://www.doczj.com/doc/916727035.html,告知。 2、由于我们的学识、经验有限,本篇难免会存在一些错误及缺陷,敬请不吝赐教:请到基因酷论坛(https://www.doczj.com/doc/916727035.html,/bbs)本篇对应的专题跟贴指出或Email genecool@https://www.doczj.com/doc/916727035.html,。 致谢: 整编者:flashhyh 主要参考资料:《生物信息学札记》樊龙江;《分子进化分析与相关软件的应用》作者不详;《进化树构建》ZHAO Yangguo;《如何用MEGA 3.1构建进化树》作者不详;《MEGA3指南》作者不详; 分子进化的研究方法 分子进化的研究方法 分子进化的研究方法 分子进化研究的意义 自20世纪中叶,随着分子生物学的不断发展,进化研究也进入了分子进化(molecularevolution)研究水平,并建立了一套依赖于核酸、蛋白质序列信息的理论和方法。随着基因组测序计划的实施,基因组的巨量信息对若干生物领域重大问题的研究提
供了有力的帮助,分子进化研究再次成为生命科学中最引人注目的领域之一。这些重大问题包括:遗传密码的起源、基因组结构的形成与演化、进化的动力、生物进化等等。分子进化研究目前更多地是集中在分子序列上,但随着越来越多生物基因组的测序完成,从基因组水平上探索进化奥秘,将开创进化研究的新天地。 分子进化研究最根本的目的就是从物种的一些分子特性出发,从而了解物种之间的生物系统发生的关系。通过核酸、蛋白质序列同源性的比较进而了解基因的进化以及生物系统发生的内在规律。 分子进化研究的基础 假设假设::核苷酸和氨基酸序列中含有生物进化历史的全部信息核苷酸和氨基酸序列中含有生物进化历史的全部信息。。 分子钟理论:在各种不同的发育谱系及足够大的进化时间尺度中,许多序列的进化速率几乎是恒定不变的。如下图: 直系同源与旁系同源 直系同源(orthologs):同源的基因是由于共同的祖先基因进化而产生的; 旁系同源(paralogs):同源的基因是由于基因复制产生的。 两者之间的关系如下图所示: 注:用于分子进化分析中的序列必须是直系同源的用于分子进化分析中的序列必须是直系同源的 用于分子进化分析中的序列必须是直系同源的,才能真实反映进化过程。 分子进化研究的基本方法 对于进化研究,主要通过构建系统发育过程有助于通过物种间隐含的种系关系揭示进化动力的实质。 表型的(phenetic)和遗传的(cladistic)数据有着明显差异。Sneath 和Sokal(1973)将表型性关系定义为根据物体一组表型性状所获得的相似性,而遗传性关系含有祖先的信息,因而可用于研究进化的途径。这两种关系可用于系统进化树(phylogenetictree)或树状图(dendrogram)来表示。表型分枝图(phenogram)和进化分枝图(cladogram)两个术语已用于表示分别根据表型性的和遗传性的关系所建立的关系树。进化分枝图可以显示事件或类群间的进化时间,而表型分枝图则不需要时间概念。文献中,更多地是使用“系统进化树”一词来表示进化的途径,另外还有系统发育树、物种树(speciestree)、基因树等等一些相同或含义略有差异的名称. 系统进化树分有根(rooted)和无根(unrooted)树。有根树反映了树上物种或基因的时间顺序,而无根树只反映分类单元之间的距离而不涉及谁是谁的祖先问题。下图表示了
构建系统进化树的方法步骤 1. 建树前的准备工作 1.1 相似序列的获得——BLAST BLAST是目前常用的数据库搜索程序,它是Basic Local Alignment Search Tool的缩写,意为“基本局部相似性比对搜索工具”(Altschul et al.,1990[62];1997[63])。国际著名生物信息中心都提供基于Web的BLAST服务器。BLAST算法的基本思路是首先找出检测序列和目标序列之间相似性程度最高的片段,并作为内核向两端延伸,以找出尽可能长的相似序列片段。 首先登录到提供BLAST服务的常用网站,比如国内的CBI、美国的NCBI、欧洲的EBI和日本的DDBJ。这些网站提供的BLAST服务在界面上差不多,但所用的程序有所差异。它们都有一个大的文本框,用于粘贴需要搜索的序列。把序列以FASTA格式(即第一行为说明行,以“>”符号开始,后面是序列的名称、说明等,其中“>”是必需的,名称及说明等可以是任意形式,换行之后是序列)粘贴到那个大的文本框,选择合适的BLAST程序和数据库,就可以开始搜索了。如果是DNA序列,一般选择BLASTN搜索DNA数据库。 这里以NCBI为例。登录NCBI主页-点击BLAST-点击Nucleotide-nucleotide BLAST (blastn)-在Search文本框中粘贴检测序列-点击BLAST!-点击Format-得到result of BLAST。 BLASTN结果如何分析(参数意义): >gi|28171832|gb|AY155203.1| Nocardia sp. ATCC 49872 16S ribosomal RNA gene, complete sequence Score = 2020 bits (1019), Expect = 0.0 Identities = 1382/1497 (92%), Gaps = 8/1497 (0%) Strand = Plus / Plus Query: 1 gacgaacgctggcggcgtgcttaacacatgcaagtcgagcggaaaggccctttcgggggt 60 |||||||||||||||||||||||||||||||||||||||||| ||||||||| ||||| Sbjct: 1 gacgaacgctggcggcgtgcttaacacatgcaagtcgagcggtaaggcccttc--ggggt 58 Query: 61 actcgagcggcgaacgggtgagtaacacgtgggtaacctgccttcagctctgggataagc 120 || ||||||||||||||||||||||||||||||| | |||||| ||||||||||||| Sbjct: 59 acacgagcggcgaacgggtgagtaacacgtgggtgatctgcctcgtactctgggataagc 118 Score :指的是提交的序列和搜索出的序列之间的分值,越高说明越相似;
极为详细的建树方法,新手入门推荐 生物进化树的构建 目录 前言 (2) 一、 NCBI (6) 二、 Mega (9) 三、 DNAMAN (15) 四、DNAStar (18) 五、 Bio edit (21)
前言 1.背景资料 进化树(evolutionary tree)又名系统树(phylogenetie tree)进化树,用来表示物种间亲缘关系远近的树状结构图。在进化树中,各个分类单元(物种)依据进化关系的远近,被安放在树状图表上的不同位置。所以,进化树简单地表示生物的进化历程和亲缘关系。已发展成为多学科(包括生命科学中 的进化论、遗传学、分类学、分子生物学、生 物化学、生物物理学和生态学,又包括数学中的 概率统计、图论、计算机科学和群论)交叉形成的一个边缘领域。 归纳总结生物进化的总趋势有以下几类: ①结构上:由简单到复杂 ②生活环境上:由水生到陆生 ③进化水平上:由低等到高等 一般来说,进化树是一个二叉树。它由很多的分支和节点构成。根据位置的不同,进化树的节
点分为外部节点和内部节点,外部节点就是我们要进行分类的分类单元(物种)。而物种之间的进化关系则用节点之间的连线表示。内部节点表示进化事件发生的地方,或表示分类单元进化的祖先。在同一个进化树中,分类单元的选择应当标准一致。进化树上不同节点之间的连线称为分支,其中有一端与叶子节点相连的分支称为外枝,不与叶子节点相连的分支称为内枝。 进化树一般有两种:有根树和无根树。有根树有一个鲜明的特征,那就是它有一个唯一的根节点。这个根节点可以理解为所有其他节点的共同祖先。所以,有根树能可以准确地反映各个物种的进化顺序,从根节点进化到任何其他节点只有能有一条惟一的路径。无根树则不能直接给出根节点,无根树只反映各个不同节点之间的进化关系的远近,没有物种如何进化的过程。但是,我们可以在无根树种指派根节点,从而找出各个物种的进化路径。 无根树 有根树
16S r D N A鉴定菌株的标准操作规程
16SrDNA鉴定菌株的标准操作规程 1.适用范围 本标准规定了通过特定引物对细菌的16SrDNA片段进行PCR扩增,然后对扩增片段进行序列分析比对,快速获得细菌种属信息的操作规程。 本标准适用于未知细菌的快速种属分析,以及为细菌的生化鉴定提供指导信息。 2.方法和原理 16SrDNA鉴定是指用利用细菌16SrDNA序列测序的方法对细菌进行种属鉴定。包括细菌基因组DNA提取、16SrDNA特异引物PCR扩增、扩增产物纯化、DNA测序、序列比对等步骤。是一种快速获得细菌种属信息的方法。 细菌rRNA(核糖体RNA)按沉降系数分为3种,分别为5S、16S和23S rRNA。16S rDNA是细菌染色体上编码16S rRNA相对应的DNA序列。16S rDNA由于大小适中,约1.5Kb左右,既能体现不同菌属之间的差异,又能利用测序技术较容易地得到其序列,故被细菌学家和分类学家接受。 在 16S rRNA 分子中,可变区序列因细菌不同而异,恒定区序列基本保守,所以可利用恒定区序列设计引物,将16S rDNA片段扩增出来,利用可变区序列的差异来对不同菌属、菌种的细菌进行分类鉴定。 16SrDNA序列的前500bp序列变化较大,包含有丰富的细菌种属的特异性信息,所以对于绝大多数菌株来说,只需要第一对引物测前500bp序列即可鉴别出细菌的菌属。针对科学论文发表或是前500bp无法鉴别的情况,需要进行16SrDNA的全序列扩增和测序,得到较为全面的16SrDNA的序列信息。
由于测序仪一次反应最多只能测出700bp的有效序列,为了结果的可靠性,通常将16SrDNA全长序列分成3部分进行测序。 3.设备和材料 3.1器材 移液器(1000μL、200μL 、100μL、10μL);涡旋振荡器;Eppendorf MixMate;离心机;水浴锅;电泳仪;制冰机;低温冰箱;PCR仪:Veriti 96 Well Thermal Cycler;凝胶成像仪:VersaDoc MP 4000;基因分析仪: AB3500、AB3130 3.2试剂 DNA快速提取试剂:PrepMan Ultra;琼脂糖;PCR试剂:Taq酶,10×Taq Buffer(Mg2+),dNTPs,ddH2O等; ExoSAP-IT;测序试剂:BigDye Terminator,5×Sequencing Buffer;BigDye XTerminator Purification Kit; 3.3耗材 移液器吸头:1000μL、200μL、10μL;离心管:1.5mL、200μL;Micro Amp TM Optical 96-Well Reaction Plate;Micro Amp TM Optical Adhesive Film;