
第27卷第6期2012年12月 大学化学
UNIVERSITY CHEMISTRY Vol.27No.6
Dec.2012
卟啉的生物合成途径与化学合成方法的比较*
苏优拉1 张逸2 李嘉宾3** 陆军农3
(1中国药科大学2010届基地班本科生;2中国药科大学2010届制药工程专业本科生;3中国药科大学
无机化学教研室药学基础化学实验中心 江苏南京211198)
摘要 对卟啉的生物合成途径和化学合成方法进行简要介绍,并尝试通过比较分析,寻找它们之间的联系,以期对卟啉化学合成方法的改进提供一些有益的信息三
关键词 卟啉 化学合成 生物合成
在自然界的生命体中,有一些化合物发挥着非常重要的作用,比如:叶绿素,其介导的光合作用将光能转化为化学能储存于植物体中,是地球上有机体生存和发展的源泉;细胞色素C,能促进氢与氧的结合,加强体内的氧化供能反应,是细胞呼吸过程中电子传递体的主要组成部分;血红素,作为血红蛋白和肌红蛋白的核心结构域,负责氧气和二氧化碳的转运,在生物体的新陈代谢中起着举足轻重的作用三令人惊奇的是,这些化合物虽然在生物体中所处部位不同二所起作用迥异,但是,它们都含有一个共同的核心结构 卟啉三
卟啉是在卟吩环上拥有取代基的一类大环化合物的总称三卟吩是由4个吡咯环和4个次甲基桥联起来的大π共轭体系,其结构如图1所示三天然卟啉类化合物一般是卟吩的吡咯环上的氢被不同基团取代所形成的,例如图1中的血红素二叶绿素和细胞色素C三卟啉的化学合成方法虽然早在1935年就被首次报道,近年来也进行了一系列的改进,但仍存在产率低二产物分离困难二能合成的卟啉种类有限等缺点三本文介绍卟啉的生物合成途径以及近年来的一系列文献报道的化学合成方法,并尝试通过比较分析,找寻它们之间存在的联系三
图1摇重要的卟啉类化合物
1 卟啉的生物合成途径
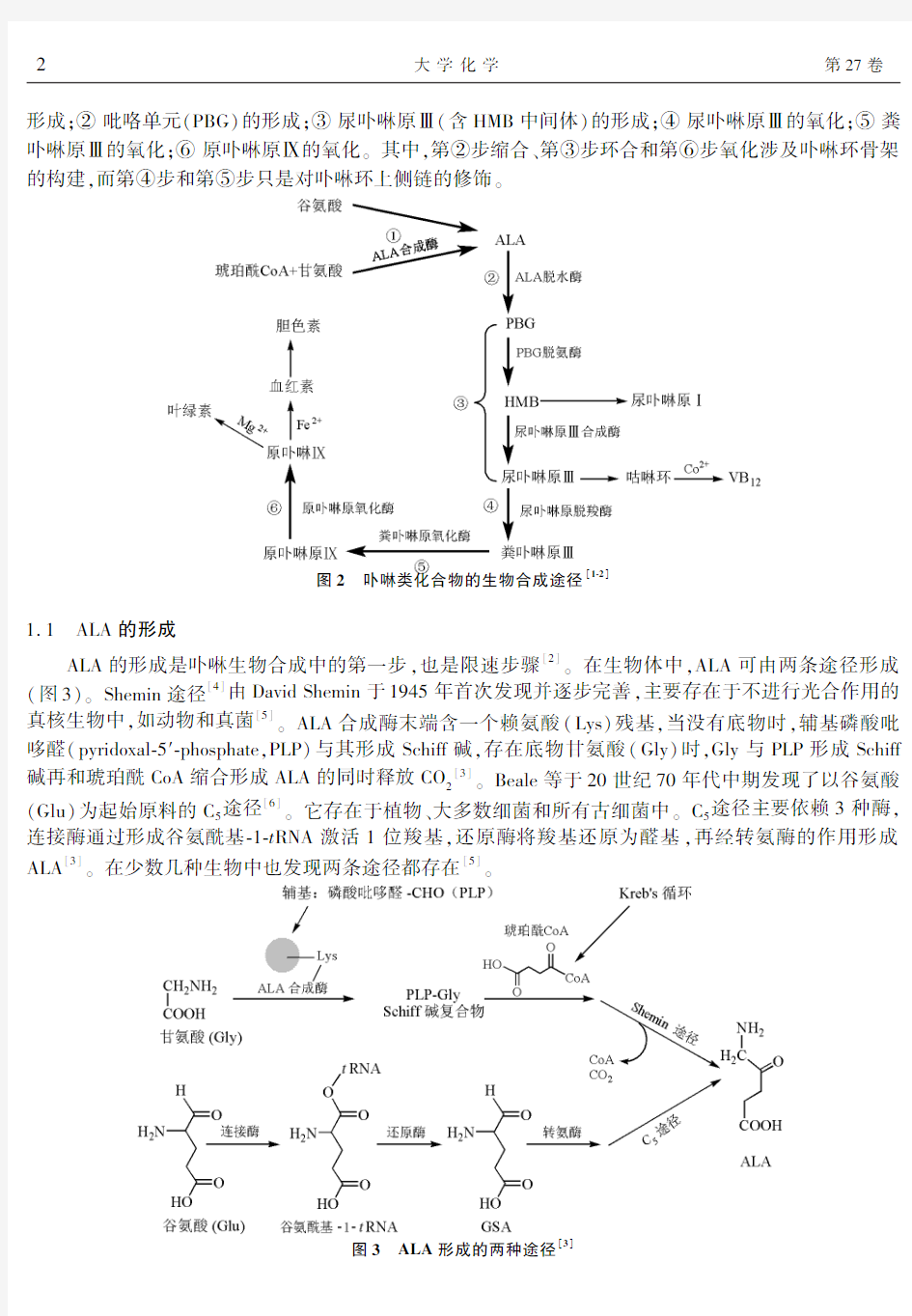
卟啉的生物合成几乎存在于所有真核细胞中,可分为6步(图2),即:①δ?氨基?γ?酮戊酸(ALA)的
* **基金资助:2009年国家大学生创新训练计划项目通讯联系人,E?mail:jbli@https://www.doczj.com/doc/8511075423.html,
2大学化学第27卷
形成;②吡咯单元(PBG)的形成;③尿卟啉原Ⅲ(含HMB中间体)的形成;④尿卟啉原Ⅲ的氧化;⑤粪卟啉原Ⅲ的氧化;⑥原卟啉原Ⅸ的氧化三其中,第②步缩合二第③步环合和第⑥步氧化涉及卟啉环骨架的构建,而第④步和第⑤步只是对卟啉环上侧链的修饰三
图2 卟啉类化合物的生物合成途径[1?2]
1.1 ALA的形成
ALA的形成是卟啉生物合成中的第一步,也是限速步骤[2]三在生物体中,ALA可由两条途径形成(图3)三Shemin途径[4]由David Shemin于1945年首次发现并逐步完善,主要存在于不进行光合作用的真核生物中,如动物和真菌[5]三ALA合成酶末端含一个赖氨酸(Lys)残基,当没有底物时,辅基磷酸吡哆醛(pyridoxal?5′?phosphate,PLP)与其形成Schiff碱,存在底物甘氨酸(Gly)时,Gly与PLP形成Schiff 碱再和琥珀酰CoA缩合形成ALA的同时释放CO2[3]三Beale等于20世纪70年代中期发现了以谷氨酸(Glu)为起始原料的C5途径[6]三它存在于植物二大多数细菌和所有古细菌中三C5途径主要依赖3种酶,连接酶通过形成谷氨酰基?1?t RNA激活1位羧基,还原酶将羧基还原为醛基,再经转氨酶的作用形成ALA[3]三在少数几种生物中也发现两条途径都存在[5]三
图3 ALA形成的两种途径[3]
1.2 PBG 的形成 两分子ALA 之间不对称缩合产生第一个吡咯衍生物 PBG(porphobilinogen)[5](图4)三反应机制与Knorr 吡咯缩合反应相似,首先,2个ALA 分子与酶活性部位的保守Lys 残基形成Schiff 碱,P 位ALA 分子的C 4和A 位ALA 分子的C 3进行Aldol 缩合形成C C 键,接着P 位ALA 分子的氨基进攻羰基碳原子形成C N 键[7]三
图4 两分子ALA 缩合形成PBG
1.3 HMB 的形成及其转化为尿卟啉原Ⅲ 4分子PBG 缩合形成尿卟啉原Ⅲ(uroporphyrinogen Ⅲ)是卟啉环生成的一个关键步骤,很多人对其具体过程提出了不同的猜想[8?10]三1980年Battersby A.R.等通过同位素标记证明了HMB(1?hydroxym?ethylbilane,羟甲基胆色烷)是尿卟啉原Ⅲ合成过程中的中间体;1987年他们又发现了新型辅基 二吡咯甲烷(dipyrromethane,DPM)[11],为阐明具体步骤奠定了基础三尿卟啉原Ⅲ
的形成由两种酶共同完图5 尿卟啉原的形成(HMB 中间体)[3,10,13]
成(图5)三首先,脱氨酶将4个PBG 组装形成开链HMB;在此过程中,先合成出二吡咯甲烷辅助因子,与脱氨酶末端的半胱氨酸巯基以硫醚键相结合,DPM 再和4分子底物相连接;当DPM 上连有4个吡咯单体时,a 环和DPM 之间的键断裂形成HMB三第二步,HMB 被转运到尿卟啉原Ⅲ合成酶上,在环合的同时,d 环重排生成尿卟啉原Ⅲ[12]三在无尿卟啉原Ⅲ合成酶时,HMB 在酸催化下迅速形成有毒性的不被代谢的尿卟啉原Ⅰ三值得注意的是,编码两种酶的基因位于同一操纵子内,两个基因协同表达[5],但
3
第6期李嘉宾等:卟啉的生物合成途径与化学合成方法的比较
尿卟啉原Ⅲ合成酶的表达量远远超过脱氨酶,以保证在生理条件下总是生成尿卟啉原Ⅲ[8]三
1.4 尿卟啉原Ⅲ 粪卟啉原Ⅲ 原卟啉原Ⅸ 尿卟啉原脱羧酶(uroporphyrinogen Ⅲdecarboxylase,UROD)和粪卟啉原氧化酶(coproporphyrinogen Ⅲoxidase,CPOs)催化卟啉环侧链的修饰(图6)三在生理底物浓度下,4个环的脱羧作用是按d →a →b →c 的顺序发生;当底物浓度超过生理浓度时,脱羧作用以随机方式发生[5]三脱羧后生成的粪卟啉原Ⅲ(coproporphyrinogen Ⅲ)在哺乳动物中,经过位于线粒体外膜的依赖O 2的CPOs 催化,最终把电子传递给O 2三此过程不需要金属和辅助因子辅助[2],CPOs 的催化机理至今仍不清楚[5],详细过程见文献
[14]三
图6 卟啉环侧链的修饰[2]
1.5 原卟啉原Ⅸ的氧化[15]
原卟啉原氧化酶(protoporphyrinogen Ⅸoxidase,PPOs)位于线粒体内膜的外表面,以O 2作为最终的电子受体,以FAD 为辅助因子,催化原卟啉原Ⅸ(protoporphyrinogen Ⅸ)的氧化,最终形成完全共轭的大环体系(图7)三生成的原卟啉Ⅸ(protoporphyrin Ⅸ)被直接转运到亚铁螯合酶上,以避免其对细胞的损害(原卟啉Ⅸ对光高度敏感,在O 2存在下,经光照射会产生自由基)三
图7 原卟啉原Ⅸ的氧化
2 卟啉的化学合成方法总结 目前,卟啉的化学合成方法主要有两种:①4个吡咯单体直接缩合环化生成卟啉(简称四吡咯合成法);②模块法三合成方法和路线的选择取决于目标卟啉分子的结构特点,中位对称取代的卟啉主要用四吡咯合成法,而不对称卟啉二天然卟啉及其类似物主要采用模块法合成三
2.1 四吡咯合成法2.1.1 Rothemund 法 卟啉类化合物最早由Rothemund 合成[16]三Rothemund 法以醛类化合物(甲醛二乙醛二苯甲醛等)和吡咯为原料,以吡啶和甲醇为溶剂在封管中反应,90~95℃下反应24~48h(图8)三该法反应时间长,所需反应条件苛刻,而且后处理非常麻烦,产率很低;在此条件下,能用来作反应物的取代苯甲醛极少[17]三
4
大学化学第27卷
图8 Rothemund 法
2.1.2 Adler?Longo 法及其改进 Adler 和Longo 等以有机质子酸作催化剂成功地制备了卟啉,并在1964年提出了卟啉生成的反应机理[18]三该法采用苯甲醛和吡咯在丙酸中回流反应30min,经冷却二过滤二洗涤及真空干燥,得到四苯基卟啉,产率达20%(图9)三此法的优点是操作比较简单,实验条件不算苛刻,产率较高三但由于反应条件的限制,一些带敏感基团的取代苯甲醛不能用作原料,带有强吸电子基的苯甲醛为底物时产率特别低;反应极易产生大量焦油状物,导致纯化非常困难;另外反应中的副产物四苯基二氢卟啉与四苯基卟啉分离较困难[17]
三
图9 Adler?Longo 法
潘继刚等[19]对Adler?Longo 法作了进一步调整,他们采用催化量的有机酸和极性溶剂代替丙酸介质,反应过程中产生的杂质明显减少,四苯基卟啉的产率最高达到50%三研究溶剂和催化剂对反应的影响,发现H +在反应过程中起催化剂的作用,p K a 2.0~4.0的酸作催化剂,合成产率较高三以二甲苯二甲苯二氯苯二硝基苯二苯甲醚为溶剂,四苯基卟啉产率较高,可达30%~50%三
2.1.3 Lindsey 法及其改进 Lindsey 等[20]进一步改进了四苯基卟啉的合成,采用苯甲醛和吡咯在氮气保护下,在二氯甲烷中,以三氟化硼和乙醚络合物催化,整个反应分两步进行,先得到卟啉合成的中间体卟啉原(porphyrinogen),然后,以二氯二腈基苯醌(DDQ)或四氯苯醌(TCQ)将卟啉原氧化得到最终产物卟啉,从而使反应可以在常温下进行三近20年来,Lindsey 小组进一步研究了此一锅两步合成法的影响因素,发现酸催化剂的种类和用量二醛和吡咯上的取代基以及反应物浓度均会影响反应产率,并对主要副产物 链状聚合物的组成进行了分析[22?24]三由于Lindsey 法的反应温度较低,较少产生焦油状副产物,目标产物的分离提纯较容易;同时较低的反应温度也允许反应物先经过化学修饰,连接上一些敏感基团,平均产率可达45%~50%(图10)三但该反应浓度低,且最大反应容积为1L,放大后效果不好[21]三反应条件较苛刻,需要无水无氧操作,且反应还不能一步生成四苯基卟啉,必须在反应过程中另外加入氧化剂三 郭灿城等[25]采用N ,N ?二甲基甲酰胺为溶剂,无水AlCl 3为催化剂,苯甲醛和吡咯缩合生成四苯基卟啉,产率可达30%,高于Adler 法三反应过程中不需氮气保护,产物不含副产物四苯基二氢卟啉,并且反应时间也较短,为2h三该方法的应用范围较广,对于以取代苯甲醛为原料的合成反应,产率在25%~30%之间三缺点是催化剂AlCl 3易与水反应,给产物的分离造成困难三
5
第6期李嘉宾等:卟啉的生物合成途径与化学合成方法的比较
6大学化学第27卷
图10 Lindsey法
Adler?Longo法和Lindsey法是目前应用比较广泛的两种方法三它们的主要区别在于所用的催化剂不同,Adler?Longo法用有机质子酸作为催化剂,而Lindsey法则用Lewis酸作为催化剂三
2.1.4 微波催化合成法
1986年,Gedye等[26]发现微波可显著加快有机合成反应速率三从此,微波在合成化学领域迅速得到重视三以二甲苯为溶剂,对硝基苯甲酸为催化剂使苯甲醛和吡咯在微波炉内反应20min,可以得到卟啉,产率为9.5%[27]三微波辅助合成卟啉在我国发展较快,研究表明微波作用的时间与强度二溶剂及催化剂的选择二反应试剂的组成及用量等均对卟啉的合成有较大的影响三此法避免了传统加热合成方法的反应时间较长(一般需回流2~3h)二副反应多二产率不高且产物难提纯等缺点,且符合节能环保二绿色化学的发展趋势[28]三
2.2 模块法
模块法主要包括[2+2]和[3+1]两种方法三
[2+2]合成亦称MacDonald方法[29],即两分子二吡咯甲烷缩合产生卟啉母核三该法可方便地合成具有C2对称轴的四苯基卟啉,也称为trans?卟啉,还可合成中位是4个不同芳基取代的卟啉三近年来,随着原料二吡咯甲烷衍生物合成方法的逐步改进和优化,可以合成的衍生物种类逐渐增多,产率也得到了提高,使[2+2]法的应用范围越来越广[30]三
[3+1]法是由MacDonald方法衍生出来的,将一个由两个桥碳原子连接的3个吡咯环组成的胆色素分子和一分子a,a′?二甲酰基吡咯环合得到卟啉的合成方法三此方法总产率较低,能合成一些结构复杂且较为特殊的卟啉,主要用于扩充卟啉的种类[31]三
关于卟啉化学合成方法更详细的总结可参考文献[32]三
3 化学合成和生物合成之间的联系
3.1 起始原料
化学合成和生物合成两种途径都以单个吡咯环作为合成卟啉环的起始原料三不同的是,化学合成通常以吡咯或取代吡咯以及醛类为原料,而生物合成途径中由两个ALA分子通过类似Knorr吡咯缩合产生的带亚甲氨基侧链的吡咯单元(PBG)作为起始原料三
3.2 单体吡咯的聚合方式
在生物合成和化学合成过程中,吡咯都是通过质子化二脱氨或脱水二再去质子的3步循环逐步加成形成聚合物三在此过程中都会产生活泼烯键,以促进吡咯的聚合(图11)三在生物合成中,吡咯单元(PBG)自身带有亚甲氨基侧链(来自甘氨酸),侧链脱氨之后形成烯键而被活化;而在化学合成中则是吡咯进攻醛基,产生带羟基的侧链,羟基质子化脱水后形成烯键而被活化三
图11 单体吡咯聚合的可能机理
(a )生物合成;(b )化学合成
3.3 链状吡咯聚合物环合形成卟啉原
两种途径都是单体吡咯先聚合成链状吡咯聚合物,然后4个吡咯单体经亚甲基桥再进一步环合成卟啉原(图12)三在生物合成途径中,PBG 脱氨酶与辅助因子DPM 和吡咯聚合物形成复合物,当连有4个吡咯时,环a 和酶之间的键会水解断裂形成链状HMB三其原因至今仍是一个谜,可能与酶的空间结构和新型辅助因子二吡咯甲烷有关;可能类似于生物体内多糖和多肽的形成过程三 根据Adler 等提出的机理,化学合成时吡咯和醛类先形成长短不一的链状聚合物,推测反应中形成的焦油状副产物很可能是吡咯和醛类的链状聚合物或者吡咯自身的聚合物三主要生成环状四聚体可能与卟啉独特的空间结构的稳定性有关三 在环合时,生物合成途径有一步特有的环翻转过程,HMB 的亚甲基被活化后,进攻与其距离较远的C 16,与C 16环合形成螺中间体,d 环翻转,碳链在另一处断裂再环合形成尿卟啉原Ⅲ三而化学合成没有环的翻转过程,活泼烯键直接与C 19环合形成卟啉环,与体内尿卟啉原Ⅰ的形成过程类似三
3.4 卟啉原氧化形成大环共轭的卟啉 在生物合成中,原卟啉原Ⅸ在PPOs 的催化下形成大环共轭的卟啉三PPOs 以二聚体形式存在,由3个结构域组成(图13),分别为FAD 结合域二底物结合域和膜结合域三原卟啉原氧化酶的辅助因子FAD 发挥重要的电子传递作用,以O 2作为最终的电子受体,因其每次只能转移两个电子,所以要经过3个独立的循环才能氧化完全,产生二氢卟啉和四氢卟啉中间体(图14)三当酶与底物结合时,底物带负电荷的丙酸基侧链与酶的精氨酸残基结合,使底物以一定方向被固定在酶上,只能以a 环和d 环间的亚甲基桥通过酶的狭缝与FAD 的N 5原子接触,环上的氢原子通过亚胺?烯胺互变异构重排并逐步被氧化三 在化学合成中,4分子醛与吡咯缩合,最终环合后产生卟啉原三1970年,Dolphin 等证实卟啉原确实是卟啉合成过程的中间体[31]三卟啉原不稳定,极易被氧化,但在一般条件下氧化又不完全,产生的二氢卟吩混在卟啉中很难除去[35]三在反应中加入氧化剂或在反应结束后再进一步氧化能提高产率三硝基
7
第6期李嘉宾等:卟啉的生物合成途径与化学合成方法的比较
图12 由吡咯单体形成卟啉原的过程
(a )生物合成;(b )
化学合成
图13 原卟啉原氧化酶的空间结构[15]
8大学化学第27卷
图14 生物合成中原卟啉原Ⅸ的氧化[18,33?34,41]
苯[36]二DMSO [37]二DDQ [20]以及最近文献报道的MnO 2[38]和SeO 2[39]等均能作为氧化剂,实现从卟啉原到卟啉的氧化,但氧化反应的具体机理至今仍不太清楚三 在卟啉原的氧化过程中,生物合成和化学合成都是通过互变异构转移H N 和H C,逐步延长共轭链,产生二氢卟啉和四氢卟啉的中间体并最终形成大环共轭的卟啉三不同的是在生物合成过程中,底物被酶包裹,由于酶对底物的固定作用,卟啉原上所有的氢必须都转移到C 20上才能被氧化;而且由于酶蛋白对底物的稳定作用,中间体二氢卟啉和四氢卟啉可以以不太稳定的形式存在三而在化学合成过程中,氧化反应可以在任一亚甲基桥上发生,而且氧化后氢转移的最终结果要保证共轭链的延伸,中间体二氢卟啉和四氢卟啉要以相对稳定的形式存在三
9
第6期李嘉宾等:卟啉的生物合成途径与化学合成方法的比较
01大学化学第27卷 4 总结和展望
近年来,由于卟啉类化合物独特的分子结构,在仿生学二药物化学二分析化学二光物理与化学二材料化学二电化学二催化化学等领域中被广泛地研究与应用,但是较低的化学合成产率限制了进一步发展三本文通过比较卟啉的生物合成途径和化学合成方法,尝试寻找二者的联系,希望能为改进化学合成方法二提高合成产率提供一些启示三
生物合成和化学合成一样,也遵循基本的化学反应规律,反应的本质是相同的三只不过由于酶的参与,酶的辅基或酶上的某个保守基团与底物相互作用,固定或增强底物的反应活性,稳定中间体的结构,降低反应的活化能,可使化学反应高效专一地进行三在化学合成中,影响产率的主要是底物的活性二对聚合度的控制以及卟啉原的氧化;而生物合成在这3方面的控制很精密三基于上述生物合成与化学合成的联系与差别,可以尝试从以下几方面对化学合成进行改进三
(1)在生物合成过程中,底物的反应活性较高三例如,吡咯聚合时,氨基被质子化后经脱氨酶作用迅速形成活泼烯键,易于被进攻三而在化学合成中,活泼烯键的形成要通过脱水,而在一般条件下自动脱水比较困难三在底物上引入其他适当的离去基团可以促进缩合,有时甚至可以省去最后一步的氧化,直接生成卟啉三Pierre Martin小组2010年报道的[2+2]法就是在底物上引入碘原子,缩合时消除碘化氢和水,不用加氧化剂直接在室温下生成大环共轭的卟啉[40]三此方法的原理若能推广至其他卟啉合成方法中,有可能显著降低反应温度,提高产率三
(2)生物合成途径对副产物的控制非常严格,原料利用率较高三吡咯聚合时一端通过巯基与酶相连,只有另一端延伸三当尿卟啉原Ⅲ合成酶催化链状吡咯聚合物的环合时,将聚合度严格控制在4,不会生成长链吡咯聚合物或者多元环状吡咯聚合物三这一点若化学合成则实现起来较困难三而且,目前的化学合成都需要酸催化,吡咯在酸性条件下容易因聚合而被破坏三抑制吡咯自身的聚合并精确控制吡咯和醛类的链状聚合物的聚合度尚有待于进一步改善三可以先在酸催化下不断延伸生成长链聚合物,将聚合物的一端固定,再用PBG脱氨酶和尿卟啉原Ⅲ合成酶从另一端进行切割和组装;或者在反应中加入具有空腔的材料,而空腔的大小恰好能容纳四聚体,这样就能把反应分割成多个单元,抑制聚合物的形成三例如,各种型号的分子筛孔径大小不同,具有一定的酸性,又能吸水,可以尝试三此外,既然卟啉能和金属离子形成配合物,可以模拟冠醚的合成方法,通过金属离子的模板效应来促进四聚体的形成三
(3)生物合成途径的最后一步为芳构化反应,在辅助因子FAD的作用下,以氧气作为最终的电子受体,实现了大π共轭体系(即卟啉环)的构建三在化学合成模拟生物合成时,聚合和氧化这两步分别进行,以避免在聚合过程中生成氧化的中间体,阻碍四聚体的形成三最近有文献报道分别以MnO2和SeO2作为氧化剂,用于卟啉原的氧化,能提高产率三它们虽然易于后处理,但都会污染环境三继续寻找价格低廉且环境友好的氧化剂,温和并高效地完成芳构化反应,符合当前绿色化学的发展趋势和要求三
参 考 文 献
[1] Scott A I.J Org Chem,2003,68(7):2529
[2] Ajioka R S,Phillips J D,Kushner J P.Biochimica et Biophysica Acta,2006,1763:723
[3] Porra R J.Photochem Photobiol,1997,65(3):492
[4] Battersby A R.Nat Prod Rep,2000,17:507
[5] Heinemann I U,Jahn M,Jahn D.Arch Biochem Biophys,2008,474:238
[6] Beale S I.Proc Natl Acad Sci USA,1975,72(7):2719
[7] Leeper F J.Nat Prod Rep,1985,2(1):19
[8] Frydman B,Frydman R B.Acc Chem Res,1975,8(6):201
[9] Scott A I,Ho K S,Kajiwara M.J Am Chem Soc,1976,98(6):1589
[10] Battersby A R,Mcdonald E.Acc Chem Res,1979,12(1):14
[11] Hart G J,Miller A D,Battersby A R,et al.J Chem Soc,Chem Commun,1987,23:1762
[12] Battersby A R.J Nat Prod,1988,51(4):629
[13] Leeper F J.Nat Prod Rep,1985,2:561
[14] Jackson A H,Elder G H,Smith S G.Int J Biochem,1978,9(12):877
[15] Koch M,Breithaupt C,Messerschmidt A,et al.EMBO J,2004,23:1720
[16] Paul R.J Am Chem Soc,1935,57:2010
[17] 郝晓伶,韩士田,刘彦钦.河北师范大学学报(自然科学版),2009,33(1):85
[18] Adler A D,Longo F R,Williams H.J Am Chem Soc,1964,84(15):3145
[19] 潘继刚,何明威,刘轻轻.有机化学,1993,13(5):533
[20] Lindsey J S,Schreiman I C,Hsu H C,et al.J Org Chem,1987,52(5):827
[21] 杨彪.精细化工,1999,16:56
[22] GeierⅢG R,Lindsey J S.J Porphyrins Phthalocyanines,2002,6:159
[23] GeierⅢG R,Lindsey J S.J Chem Soc,Perkin Trans2,2001(5):677
[24] GeierⅢG R,Lindsey J S.Tetrahedron,2004,60:11435
[25] 郭灿城,何兴涛,邹纲要.有机化学,1991,11(4):416
[26] Gedye R.Tetrahedron Lett,1986,27(3):279
[27] Petit A.Synth Commun,1992,22(8):1137
[28] 汉玉霞,韩士田,刘彦钦.化学工程与装备,2008,6:98
[29] Arsenault G P,Bullock E,Macdonald S F.J Am Chem Soc,1960,82:4384
[30] Lindsey J S.Acc Chem Res,2010,43(2):300
[31] 王周锋,邓文礼.化学进展,2007,19(4):520
[32] akthitharan S,Edwards C,Boyle R W.Tetrahedron,2000,56:1025
[33] Massey V.Biochem Soc Trans,2000,28(4):283
[34] Mattevi A.Trends Biochem Sci,2006,31:276
[35] 王君文,何明威.化学试剂,2001,23(1):9
[36] 杨琴,冯清.中国药物化学杂志,2006,16(3):154
[37] 章艳,高保娇.合成化学,2008,16(1):86
[38] Bruno F O N,António M R G,Marta P.Inorg Chem Commun,2010,13:395
[39] Stephanie M S L,Diogo R B D,Eugênia R D,et al.Tetrahedron Lett,2011,52:1441
[40] Pierre M,Markus M,Dietmar F,et https://www.doczj.com/doc/8511075423.html, Process Res Dev,2010,14:799
[41] Banerjee R.REDOX BIOCHEMISTRY.Hoboken,New Jersey:Wiley John&Sons,Inc,200811
第6期李嘉宾等:卟啉的生物合成途径与化学合成方法的比较
各类中药化学成分的主要生物合成途径 乙酸-丙二酸途径:脂肪酸类,酚类,醌类;甲戊二羟酸途径:萜类,甾类;莽草酸途径:即桂皮酸途径,苯丙素类,木脂素类,香豆素类;氨基酸途径 :生物碱类 溶剂提取法(常用溶剂及极性) (1)溶剂按极性分类:三类,即亲脂性有机溶剂、亲水性有机溶剂和水。溶剂按极性由弱到强的顺序如下:石油醚<四氯化碳<苯<二氯甲烷<氯仿<乙醚<乙酸乙酯<正丁醇<丙酮<甲醇(乙醇)<水。 甲醇(乙醇)是最常用的溶剂,能用水任意比例混合. 分子大,C多,极性小,反之,大..按相似相溶原理,极性大的溶剂提取极性大的化合物 提取方法 ①煎煮法:挥发性及加热易破坏,多糖类不宜用。 ②浸渍法:不用加热,适用于遇热易破坏或挥发性成分,含淀粉或黏液质多的成分,但效率不高。 ③渗漉法:效率较高。④回流提取法:受热易破坏的成分不宜用。⑤连续回流提取法:有机溶剂,索氏提取器或连续回流装置。⑥水蒸气蒸馏法: 适于具挥发性,能随水蒸气蒸馏而不被破坏的。挥发油、小分子生物碱、酚类、游离醌类等:⑥超临界萃取法:以CO2为溶剂.用于极性低的化合物,室温下工作,几乎不用有机溶剂,环保 分离方法 ①吸附色谱:利用吸附剂对被分离化合物分子的吸附能力的差异,而实现分离的一类色谱。硅胶用于大多数中药成分;氧化铝用于碱性或中性亲脂性成分如生物碱、萜、甾;活性炭用于水溶性物质如氨基酸、糖类和某些苷类;聚酰胺用于酚醌如黄酮、蒽醌及鞣质。②凝胶色谱:主要是分子筛作用,根据凝胶的孔径和被分离化合物分子的大小而达到分离目的。③离子交换色谱:基于各成分解离度的不同而分离。主要用于生物碱、有机酸及氨基酸、蛋白质、多糖等水溶性成分的分离纯化。④大孔树脂色谱:一类没有可解离基团,具有多孔结构,不溶于水的固体高分子物质。它可以通过物理吸附有选择地吸附有机物质而达到分离的目的。是反相的性质,一般被分离物质极性越大,越先被洗脱下来,极性越小,越后洗脱下来。应用于中药有效部位或有效成分的分离富集。⑤分配色谱:利用物质在固定相和流动相之间分配系数不同而达到分离。正相色谱:固定相极性>流动相极性,用于分离极性和中等极性的成分。常用固定相:氰基或氨基键合相;常用流动相为有机溶剂。反相色谱:固定相极性<流动相极性,用于离非极性和中等极性的成分,常用C18或C8键合相。常用流动相为甲醇-水或乙腈-水。 糖和苷类化合物 糖:多羟基醛或多羟基酮及其衍生物、聚合物的总称 苷:糖或糖额衍生物与另一非糖物质通过糖的端基碳原子连接而成,又称配糖体 构型D,L,α,β : 向上D,向下L; 同侧:β异侧:α 苷键酸水解:苷键原子首先发生质子化,然后苷键断裂生成苷元和糖的阳碳离子中间体,在水中阳碳离子经溶剂化,再脱去氢离子形成糖分子。难易顺序:N-苷>O-苷>S-苷>C-苷。强酸水解:得到糖,苷元易破坏;弱酸水解:得到次级苷,确定糖的连接顺序;两相酸水解:保护苷元 酶水解:对难以水解或不稳定的苷,在酶水解条件温和,不会破坏苷元,可得到真正的苷元 显色反应 Molish反应:加入5%α-萘酚乙醇液,沿管壁缓慢滴入浓硫酸,在两层液面间会出现一个紫色环。又称α-萘酚反应.说明含有糖类或苷类. (但碳苷和糖醛酸例外,呈阴性.) 菲林和多伦反应:阳性,有还原糖.可以利用这两个反应来区别还原糖和非还原糖。 单糖:都是还原糖。双糖:麦芽糖、乳糖为还原糖。蔗糖为非还原糖 苷键构型的判断 糖苷的1H-NMR:成苷的端基质子H的耦合常在较低场。如:β构型J H1-H2=6~9Hz(8左右);α构型J H1-H2=2~3.5Hz (4左右) 醌类 酸性(规律) -COOH > 二个β-OH > 一个β-OH >二个α- OH > 一个α–OH 可用PH 梯度萃取分离。 其结果为①和②被5%碳酸氢钠溶液提出;③被5%碳酸钠提出;④被1%氢氧化钠提出;⑤只能被5%氢氧化钠提出 可用PH梯度萃取分离。 颜色反应 1、Feigl反应:全部醌类均阳性。碱性条件加热,紫色 2、Borntrager’s反应:也叫碱液试验,羟基蒽醌阳性。——颜色变化与OH数目及位置有关,红-紫色. 3、醋酸镁反应:含α-酚羟基或邻二酚羟基的蒽醌类阳性。 4、与活性亚甲基试剂反应kesting-Craven和无色亚甲蓝显色反应: 苯醌和萘醌类的专属反应.在碱性条件下 5、对亚硝基-二甲苯胺反应: 蒽酮类的特异性反 应.(唯一).蒽酮就是9或10位没有被取代的羟基 蒽酮类. 醌类化合物的提取与分离 (大题,看书) pH梯度萃取法P82 例:大黄蒽醌苷类的分离 苯丙素类(一个或几个C6-C3) 香豆素:一般具有苯骈α-吡喃酮母核的天然产物 母核(画) 内酯性质和碱水解反应 碱性开环,酸性闭环。但长时间加热,异构化,不可 恢复闭环. 显色反应有荧光性质 1、Gibb’s反应: 试剂:2,6-二氯(溴)苯醌氯 亚胺 C6位没取代,阳性,蓝色 2、Emerson反应试剂:4-氨基安替比林,铁氰化 钾反应 C6位没取代,阳性,红色 木脂素鉴识 Labat反应:具有亚甲二氧基的木脂素加浓硫酸 后,再加没食子酸,可产生蓝绿色 黄酮(C6-C3-C6) 结构与基本骨架(芦丁,槲皮素,鼠李糖,葡萄糖的 结构都要求会写)138页 经典结构是2-苯基色原酮,现在泛指两个苯环通 过三个碳原子相互连接而成的一类化合物 黄酮类:以2-苯基色原酮为母核,且3位上无含 氧基团取代的一类化合物 黄酮醇:在黄酮基本母核的3位上连有羟基或含 氧基团 二氢黄酮:黄酮基本母核的2、3位双键被氢化而 成 二氢黄酮醇:黄酮醇类的2、3位被氢化的基本母 核 交叉共轭体系:黄酮结构中色原酮部分本身无 色,但在2位上引入苯环后,即形成交叉共轭体 系,通过电子转移、重排,使共轭链延长而显出 颜色。在7位或4’位上引入-OH及-OCH3等助色 团后,产生p-π共轭,使化合物颜色加深。 溶解度:游离黄酮一般难溶于水,易溶于甲醇、 乙醇、乙酸乙酯、氯仿、乙醚等有机溶剂及稀碱 水中。引入羟基增多,水溶性增大,脂溶性降 低;而羟基被甲基化后,脂溶性增加。黄酮苷一 般易溶于水、甲醇、乙醇等强极性溶剂中,但难 溶于苯、氯仿、乙醚等有机溶剂中 平面型如黄酮、黄酮醇、查尔酮等溶解度较小, 非平面型如二氢黄酮及二氢黄酮醇的溶解性较 大,异黄酮的也较大 酸性:7,4’-二OH黄酮>7-或4’-OH黄酮>一 般酚羟基>5-OH黄酮 显色反应:(1)HCl-Mg反应:样品溶于甲醇或乙 醇1ml中,加入少许Mg,再加几滴浓HCl,一两 分钟显红~紫红色。(2)AlCl3反应:样品的乙醇 溶液和1%乙醇溶液AlCl3反应,生成黄色络合 物。(3)锆盐-枸橼酸反应:可鉴别黄酮类化合 物是否纯在3-或5-OH。样品的甲醇溶液加2%二氯 氧锆甲醇溶液。黄色不褪,有3-OH或3,5-OH, 如果减褪,无3-OH而有5-OH pH梯度萃取法:5%NaHCO3可萃取7,4’-二羟基 黄酮,5%NaCO3可萃取7-或4‘-羟基黄酮, 2%NaOH可萃取一般酚羟基的黄酮,4%NaOH可以萃 取5-羟基黄酮。 柱色谱分离 硅胶柱:利用极性差异,几乎适用于任何类型黄 酮(主要分离异黄酮、二氢黄酮,二氢黄酮醇及 高度驾机皇或乙酰化的黄酮及黄酮醇) 聚酰胺柱:通过酰胺羰基与黄酮类化合物分子上 的酚羟基形成氢键缔合而产生。化合物结构与Rf 值:酚羟基少>多;易形成分子内氢键>难;芳 香化程度低>高;异黄酮>二氢黄酮醇>黄酮> 黄酮醇;游离黄铜>单糖苷>双糖苷>叁糖苷 (含水移动相做洗脱剂);有机溶剂做洗脱剂反 之。洗脱能力由弱至强;水<甲醇或乙醇(浓度 由低到高)<丙酮<稀氢氧化钠水溶液或氨水< 甲酰胺<二甲基甲酰胺<尿素水溶液 紫外 黄酮类型带II(弱峰) 带I(强峰) 取代) 黄酮醇(3-OH 游离) 250-280 358-385 异黄酮245-270 310-330肩峰 二氢黄酮/醇370-295 300-330 查耳酮220-270低强度340-390 氢谱: 黄酮或黄酮类H-3是一个尖锐的单峰出现在 6.3 处 邻位耦合:耦合常数为8Hz左右 间位耦合:2-3Hz 对位耦合:很弱,数值很小或没有 5,7-二OH黄酮δppm:H-6小于 H-8 . 7- OH 黄酮: δppm:H-6 > H-8 6’δ比较大,5’较小 同时还要看 单峰S,就没有邻,间位双锋d说明有邻位或间位 其中一个双双锋dd就说明有邻,和间两个 生物合成途径 经验异戊二烯法则:基本碳架均是由异戊二烯以 头-尾顺序或非头-尾顺序相连而成;生源异戊二 烯法则:甲戊二羟酸是各种萜类化合物生物合成 的关键前体 单萜:无环,单环,双环,三环,环烯醚。知道 卓酚酮,环烯醚萜,薄荷醇,青蒿素的二级结构 和性质 性质:萜类多具苦味,单萜及倍半萜可随水蒸气 蒸馏,其沸点随其结构中的C5单位数、双键数、 含氧基团数的升高而规律性升高 提取:挥发性萜可用水蒸气蒸馏法;一般萜可用 甲醇或乙醇提取;萜内酯可先用提取萜的方法提 取出总萜,然后利用内酯的特性,用碱水提取酸 化沉淀的方法纯化;萜苷多用甲醇、乙醇或水提 取 柱色谱:吸附剂多用硅胶。中性氧化铝。含双键 者可用硝酸银络合柱色谱分离(利用硝酸银可与 双键形成π络合物,而双键数目位置及立体构型 不同的萜在络合程度及络合稳定性方面有一定差 异)。洗脱剂多以石油醚、正己烷、环己烷分离 萜烯,或混以不同比例的乙酸乙酯分离含氧萜 鉴识:卓酚酮类的检识 (硫酸铜反应:绿色结 晶);环烯醚萜的检识(Weiggering法:蓝色/紫红 色;Shear反应:黄变棕变深绿);薁类的检识 (Ehrlich反应:蓝紫绿;对-二甲胺基苯甲醛) 挥发油 也称精油,是存在于植物体内的一类具有挥发 性、可随水蒸气蒸馏、与水不相容的油状液体。 分为:芳香族,萜类,脂肪族 检识:化学测定常数:酸值、酯值、皂化值 提取方法:①蒸馏法:提取挥发油最常用的方 法,对热不稳定的挥发油不能用。②溶剂萃取 法:脂溶性杂质较多。③吸收法:油脂吸收法, 用于提取贵重挥发油。④压榨法:该方法可保持 挥发油的原有新鲜香味,但可能溶出原料中的不 挥发性物质。⑤二氧化碳超临界流体萃取法:有 防止氧化热解及提高品质的突出优点,用于提取 芳香挥发油 三萜 醋酐-浓硫酸反应(Liebermann-Burchard) 红-紫-蓝-绿色-褪色(甾体皂苷) 黄-红-紫-蓝-褪色(三萜皂苷) 胆甾醇沉淀法:胆甾醇复合物——乙醚回流提 取,去除胆甾醇,得皂苷。因为甾体皂苷比三萜 皂苷形成的复合物稳定. 甾类 C21甾醇C2H5 昆虫变态激素8-10个碳的脂肪烃 强心苷不饱和内酯环 甾体母核的C-17位上均连一个不饱和内酯环。根 据内酯环的不同:五元不饱和内酯环叫甲型强心 苷元;六元不饱和内酯环叫乙型。 苷和糖连接的顺序分: I型强心苷:苷元-(2,6-二去氧糖)x-(D-葡萄
Synthesis of p -substituted tetraphenylporphyrins and corresponding ferric complexes with mixed-solvents method Zhicheng SUN 1,Yuanbin SHE (?)1,Rugang ZHONG 2 1Institute of Green Chemistry and Fine Chemicals,Beijing University of Technology,Beijing 100124,China 2College of Life Science &Bioengineering,Beijing University of Technology,Beijing 100124,China ?Higher Education Press and Springer-Verlag 2009 Abstract By using mixed-solvents method,?ve kinds of p -substituted tetraphenylporphyrin compounds [T(p -R)PPH 2,R =NO 2,Cl,CH 3,OCH 3,OH]were synthesized by the condensation of p -substituted benzaldehyde with pyrrole in mixed solvents (propionic acid,acetic acid and nitrobenzene),and corresponding ferric complexes [T(p -R)PPFe III Cl]were synthesized in dimethylformamide.The above free base porphyrins were obtained in 30%–50%yields,metalation yields were up to 90%and total yields of ferric complexes were 27%–50%.Effects of reactive conditions,solvents and oxidants on yields of free base porphyrins were investigated and the relevant mechanism was discussed.Structures of the above porphyrin complexes were characterized by ultraviolet-visible (UV-Vis),infrared (IR)and far infrared (FIR)spectroscopy. Keywords porphyrin,metalloporphyrin,mixed-solvents,synthesis,characterization 1Introduction Substituted tetraphenylporphyrin complexes with conju-gated macrocycles have been essential to the study of biomimetic chemistry in recent years [1–5].The porphyrin iron complexes are mostly used for the models of cytochrome P-450in which the dioxygen has been activated by metalloporphyrins under mild conditions [6,7].Based on that,the substituted metalloporphyrins present high catalytic activities and high selectivities in the catalytic oxidation of hydrocarbons without co-reducing reagents.So the catalytic effect of metalloporphyrins on the activity of inert C-H bonds has been given considerable attention [8]. However,the yields of substituted tetraphenylporphyrin complexes are lower and the cost of synthesis is still expensive,which have exceedingly restricted their current applications.Herein,the study on ef ?cient synthesis methods for improving the yields of metalloporphyrin complexes is obviously necessary. Chemists have developed a few synthetic methods to provide convenient access to synthesize substituent tetraphenylporphyrin complexes [9–11].The prevalent method of synthesis involves a mixed aldehyde condensa-tion with pyrrole via Adler method in re ?uxing propionic acid [12].Nevertheless,several limitations remain on the scope of synthetic porphyrin chemistry.One of these is the synthesis of porphyrins with only one solvent, e.g.,propionic acid or dimethylformamide,which brings the problems of a higher boiling point and inconsistent polarity [13].Therefore,the porphyrin complexes are often with low yields and the synthetic method is not universal for porphyrin complexes with various substituents. In this paper,a series of para -substituted tetraphenyl-porphyrin compounds and the ferric complexes [T(p -R)PPFe III Cl]were synthesized by using mixed-solvents method (Scheme 1).Different reaction conditions were investigated and the yields of porphyrin complexes were improved remarkably.This approach proved to be effective for the synthesis of a varity of metalloporphyrins. 2 Experimental 2.1 Reagent and instrument All chemicals were obtained commercially and used as received unless otherwise noted.Pyrrole was redistilled before use.Dichloromethane was dehydrated.Neutral Al 2O 3was baked at 100°C for 5h. Ultraviolet-visible (UV-Vis)spectra were obtained on HITACHI U-3010.Infrared (IR)spectra were obtained on Received September 18,2008;accepted November 10,2008E-mail:sheyb@https://www.doczj.com/doc/8511075423.html, Front.Chem.Eng.China 2009,3(4):457–461DOI 10.1007/s11705-009-0169-6
卟啉化合物的合成及物理化学性质 周彬 ,张文 ,曾琪 ,张智 (武汉大学 化学与分子科学学院 ,武汉 430072) 【摘要】利用中位-四[对羟基苯基]卟啉和四水合乙酸钴在DMF 中搅拌加热至100℃回流30min 合成了金属钴卟啉。然后再用柱层析分离得到纯净的金属卟啉产物。利用电导率仪研究了金属卟啉金属钴卟啉的电迁移性质。通过金属钴卟啉配合物与咪唑配位动力学的研究证实了其轴向上存在配位作用。 【关键词】 卟啉、金属(钴)卟啉配合物、咪唑、动力学性质、电迁移性质 【前言】 卟啉化合物是一类含氮杂环的共轭化合物,其中环上的各原子处于同一平面内(如图1所示) : NH N HN N NH N HN N X X X X 图1 X=COOH;OH;NH 2 如图2
卟啉环中含有四个吡咯环,每两个吡咯环在2位与5位之间由一个次甲基桥连,在5,10,15,20,位上也可键合四个取代苯基(如图2),形成四取代苯基卟啉。卟啉环中有交替的单键和双键,有18个π电子组成的共轭体系,具有芳香性。 当两个氮原子上的质子电离后,其形成的空腔中可以容纳Fe,Co,Mg,Cu,Zn,等金属离子而形成金属配合物,并且这些金属配合物都具有一些生理上的作用。 卟啉化合物具有对光,热的良好稳定性。它的这种稳定性,大的可见光消光系数和它在电荷转移过程中的特殊作用,使得它在光电领域中的应用受到高度重视,它被用于气体传感器,太阳能的贮存,生物模拟氧化反应的催化剂,生物大分子探针,还可以作为模拟天然产物的母体,金属卟啉配合物被广泛的应用于微量分析等领域。本实验合成并提纯了卟啉配合物,采用电导仪测定金属配合物在溶液中的电迁移性质,还就其与有机碱的轴向配位反应进行动力学的测定。 【实验部分】 ⒈试剂与仪器: 1.1试剂 卟啉,醋酸钴,DMF(二甲基甲酰胺),无水乙醇,无水乙醚,二氯甲烷,丙酮,环己烷,薄层层析硅胶,柱层析硅胶,氢氧化钠,咪唑, 1.2仪器 紫外-可见分光光度仪,傅立叶变换红外光谱仪,DD3001电导率仪,分析天平,电磁搅拌器,减压蒸馏装置,旋转蒸发仪,抽滤装置,真空干燥器. ⒉实验步骤:
【摘要】本文主要介绍了利用一种用1,4-二甲氧基苯作为反应的起始原料,用氢气作为还原剂,在金属钯复合催化剂的作用下反应直接生成产物对苯二酚。此工艺简单方便易行,副产物少,反应条件相对比较温和。本文对反应的催化剂的种类进行了帅选并且对催化剂的用量、反应温度、反应压力和反应时间进行了优化,最终优化的结果可以使得对苯二酚的产率达到90%。 【关键词】 1,4-二甲氧基苯对苯二酚氢气 对苯二酚是一个重要的有机化工原料,用途非常广泛。酚主要用于制取黑白显影剂、蒽醌染料和偶氮染料、合成气脱硫工艺的催化剂、橡胶和塑料的防老剂单体阻聚剂、食品及涂料清漆、橡胶和汽油的稳定剂和抗氧化剂、石油抗凝剂、洗涤剂的缓蚀剂、稳定剂和抗氧剂等,还用于化妆品的染发剂。 目前世界上生产对苯二酚的方法主要分为以下四种(1)苯胺氧化法;(2)对二异丙苯氧化法;(3)苯酚丙酮法;(4)苯酚羟基化法。 路线1:苯胺氧化法。 目前我国大部分生产厂家仍沿用苯胺氧化法,这是对苯二酚最早的生产方法,至今已有70多年的历史。该法反应过程为:在硫酸中(将138g的1,4-二甲氧基苯和5%不同的催化剂加入烧瓶中,往体系中加入氢气,在压力10mpa和120℃的温度下反应,取样分析对苯二酚的产率。结果如表3所示。 从上表可以看出一共四种催化剂,pd/sio2-al2o3和pd/al2o3的催化效果基本上没有什么差别,分别为81%和80%,但是在产率上都低于催化剂pd/caco3和pd/deloxan apii。pd/caco3 和pd/deloxan apii的催化效果都非常好。下面对催化剂的用量进行了一些实验,结果如表4所示。 从实验结果看出,随着催化剂用量的增加,产率得到了提高,但当用量达到5%的时候,再增加用量,产率基本上没有变化,使用6%pd/caco3为催化剂的产品最终产率为96%,使用6%pd/deloxan apii为催化剂的最终产率为93%。 2.4 反应时间对反应的影响 将138g的1,4-二甲氧基苯和5% pd/caco3催化剂加入烧瓶中,往体系中加入氢气,在压力10mpa和120℃的温度下反应,取样分析对苯二酚的产率,研究反应时间对产率的影响。结果如表5所示。 从上表可以看出反应时间在2小时以下,随着时间的推移对苯二酚的产率渐渐的提高,当反应时间大于2小时的时候,对苯二酚的产率基本没有什么变化,所以反应时间规定在2小时。 3 结语 本文比较了不同种类的催化剂对此反应的影响,确定以pd/caco3或pd/deloxan apii 为反应的催化剂,并且经过对pd/caco3和pd/deloxan apii的用量进行对比实验,确定pd/caco3 和pd/deloxan apii的用量比为5%,并对温度、压力和反应时间进行了对比。最终确定最佳工艺条件为:1. pd/caco3的用量比为5%;2.反应的温度为120℃;3.反应的压力为10mpa;4.反应的时间2小时。
杜仲中的次生代谢物及其生物合成途径摘要:本文介绍了杜仲的生物学特征及产地,对杜仲中所含的次生代谢产物及其生物合成途径进行了综述。 关键词:杜仲;次生代谢产物;生物合成途径 1 杜仲概述 杜仲(Eucommia ulmoides Oliver)为杜仲科杜仲属植物,是我国特有名贵药用树种[1],落叶乔木,其高达20m、树皮灰褐色,粗糙,连同枝、叶、根均含胶,折断有银白色细丝。叶椭圆或椭圆伏卵型,长6~18cm,边缘有锯齿,下边脉上有毛,叶柄长1~2cm,果为翅果扁平而薄,内含一种子[2-3]。 杜仲为地质史上第三纪冰川运动残留下来的古生物树种,为国家二级保护植物[4],原产于我国西南诸省山区,喜温暖而凉爽的气候,属喜光树种,在强光、全光条件下才能良好生长。杜仲适生范围较广,我国有丰富的资源,主要分布于甘、陕、晋、豫、湘、鄂、川、滇、黔、桂、苏、皖、浙、赣等省、自治区,垂直分布一般在200~1500m之间,个别地区海拔高度可达2500m,其野生的分布中心是在中国中部地区[5]。在日本、俄罗斯、朝鲜、北欧、北美等国家和地区也有引种[6]。 其皮和叶是我国传统的中药材,具有补肝肾、强筋骨和安胎的作用,用于治疗肾虚腰痛、筋骨无力、胎动不安、高血压、头晕目眩等症。杜仲皮中主要药用成分为松脂醇二葡萄糖苷,杜仲叶中主要药用成分为绿原酸[7]。 2 杜仲的化学成分 , 近年来,各国学者对杜仲的化学成分进行了大量研究,目前经过分离和鉴定的有机化合物约有70种以上,无机矿物元素不少于15 种。研究还发现,杜仲皮、花、叶和枝条等各部分中含有相似的化学成分,主要包括: 苯丙素类、木脂素类、环烯醚萜类、黄酮类、多糖、氨基酸和杜仲胶等有机化合物,及钙、铁等
步骤缺点备注 Rothemunde 法以荃类和吡咯为原料,以吡啶 和甲醇为溶剂。在封口的玻璃 管中反应,水浴90—95度下 反应30个小时。将反应液降 温后过滤,以吡啶洗涤反应管 和虑饼,合成虑液,再以百分 之五十乙酸萃取两次。最后将 醚液用饱和NAHSO3萃取三 次后,水洗至中性 反应时间长,反应条件苛刻,且要 求反应器密闭,底物浓度较低,后 处理非常麻烦,反应收率低 Adler-longo 法苯甲醛和新蒸的吡咯在丙酸 中回流30min。冷却至室温后 过滤,然后分别用甲醇和热水 洗涤滤饼,得到蓝紫色晶体, 最后真空干燥。 由于反应条件的限制,一些带敏感 基团或对酸敏感的取代苯甲醛不 能用作原料,同时带有强吸电基的 苯甲醛进行合成时产率特别低,而 且由于底物浓度大以及反应的温 度高,在反应过程中容易长生大量 的焦油,产物不容易纯化。 Lindsey法在室温下采用苯甲醛和吡咯 为原料,在氮气保护下,以二 氮甲烷为溶剂,三氟化硼乙醚 络合物为催化剂,生成卟啉 原,然后以二氯二氰基苯醌将 四苯基卟啉原氧化得到最终 产物四苯基卟啉,收率可达 20—30 优点:反应条件温和,不会产生焦油状的副产物,且产率较高,适合合成带有敏感基团或是空间位阻较大的卟啉。 缺点:此反应只能在比较稀的溶液中进行,且反应步骤相对较多。不仅原料较为昂贵,且反应过程需要无水及无氧操作 [2+2]法利用两分子的二吡咯甲烷缩 合成卟啉优点:可以方便的合成出各种带有不同取代基的不对称的卟啉,且产率比较高,具有较强的灵活性和区域选择性 缺点:合成过程中消耗会比较大且这类反应要在酸性条件下催化进行,而在该条件下容易使得二吡咯甲烷裂解,从而不利于反应的进行。同时,吡咯也容易进行自身缩合反应,且缩合产物难于分离。 微波激励法将吡咯和苯甲醛附于无机载 体硅胶上,利用载体的酸性催 化作用,在微波激励下合成四 苯基卟啉,反应10min后, 直接加入层吸柱进行吸分离, 得到四苯基卟啉,收率百分之 9.5 以二甲苯为溶剂,对硝基苯甲酸为催化剂,使苯甲醛吡咯在微波炉中反应20min,收率可达到百分之42.
2012.11.13-2012.11.22 卟啉化合物的合成、理化性质及其应用 姓名(学号) 苏州大学材料与化学化工学部09级化学专业 摘要:本实验采用在DMF溶液中缓慢滴加等摩尔比的吡咯和苯甲醛混合液,油浴加热反应,在经结晶过柱旋转蒸发得到纯产品四苯基卟啉(TPPH2)。 关键词:卟啉、制备、金属卟啉 Abstract:This experiment in the DMF solution such as slow drop and mole ratio of pyrrole and benzaldehyde mixture, oil bath heating reaction, the crystallization in a column rotary evaporation get pure product four phenyl porphyrin (TPPH2). Keyword:porphyrin、preparation、metalloporphyrin 1.前言 卟啉化合物是一类含氮杂环的共轭化合物,其中环上各原子处于同一平面内。在植物中的叶绿素、红血球中的血红蛋白、肌肉中的肌红蛋白、动物的肝脏、血液细胞、植物中的过氧化氢酶、牛奶等一系列具有重要生理功能的物质中,都含有卟啉或类卟啉的骨架。它们都是起着重要的生理作用的活性中心。除了生物活性外,卟啉及类卟啉化合物具有大共轭平面的特殊结构,使得其广泛应用于催化、新材料的开发、微量分析等领域。 本实验采用在DMF溶液中缓慢滴加等摩尔比的吡咯和苯甲醛混合液,油浴加热反应,在经结晶过柱旋转蒸发得到纯产品四苯基卟啉(TPPH2)。 2.实验部分 2.1、仪器与药品 仪器:烧杯(50mL×2、100mL×1)、量筒(50mL)、三颈烧瓶(250mL,19#×1、14#×2)、双颈烧瓶(50mL,19#×2)、茄形烧瓶(250mL,24#)、滴液漏斗(14#)、球形冷凝管(19#)干燥管(19#)、空心塞(19#×2、14#×2)、布氏漏斗及抽滤瓶、调压变压器、旋转蒸发仪、温度计(300℃)、氩气钢瓶、干燥器、油浴、磁力搅拌器、回流装置。 药品:DMF、无水氯化铝、吡咯、苯甲醛、乙醇、中性氧化铝、二氯甲烷、
CoP-CMP的合成: 在50ml三口烧瓶中加入158.5mg对溴苯基钴卟啉、63.6mg1,4-苯二硼酸、 K 2CO 3 (221.1mg)溶液6ml、1,4-二氧六环30ml、Pd(PPh3) 4 18.5mg。冷冻到-70℃ 后通过三次15min-泵抽15min-通氮气-解冻除氧。然后在90℃下反应24小时。反应24小时后过滤,用去离子水、无水乙醇、THF、丙酮洗涤至滤液无色,再用甲醇、THF、丙酮索氏提取。最终得到0.1302g棕色粉末。 产物中的白色杂质比上次做的少很多,白色杂质可能是三口烧瓶的玻璃被碱腐蚀掉进产物里。由于对溴苯基钴卟啉在溶剂里溶解度低,产物很难洗干净。 对四硝基苯基卟啉(TNPP)的合成: 方法一:在装有冷凝管的250ml三口烧瓶中加入3.67g(24mmol)对硝基苯甲醛、3.9ml(41mmol)醋酸酐和100ml丙酸,加热至回流。10min内滴加1.6102g (48mmol)吡咯(溶于3ml丙酸),然后继续反应30min。反应混合物冷却至室温后过滤,得到的黑色固体用去离子水洗涤后40℃下真空干燥。得到的黑色固体里含有大量聚合物,这些聚合物易溶解于吡啶,而产物在冷的吡啶溶液里溶解度低。把黑色固体在50ml吡啶中回流1小时,冷却至室温后放在冰箱里过夜。混合物过滤后用丙酮洗涤固体至滤液无色,最后干燥得到0.8034g紫色产物。产率16.85%。 方法二:在装有冷凝管的100ml三口烧瓶中加入4g对硝基苯甲醛、7ml乳酸和25ml硝基苯,加热至130℃。20min内滴加12ml溶有1.71g吡咯的硝基苯溶液后继续反应2个小时。然后在60℃下加入20ml甲醇,搅拌30min。把反应物放在冰箱里过夜后抽滤得到紫黑色固体。然后把紫黑色固体在10ml吡啶中回流1个小时后放在冰箱里过夜。最后抽滤、用丙酮洗涤固体至滤液无色,最后干燥得到0.9735g紫色产物。产率18.5%。 UV-Vis(λ;nm;CHCl 3 溶剂):422,514,550,590,650。 对四氨基苯基卟啉(TAPP)的合成: 氮气保护下在装有冷凝管的250ml三口烧瓶中加入1.44g(108mmol)对四硝基苯基卟啉和HCl(12mol/l,70ml)。在75-80℃下加入14ml溶解有13.04 g SnCl 2·2H 2 O (58mmol)的浓盐酸至三口烧瓶中反应30min。然后在冰浴条件下加 入80ml浓氨水终止反应,在空气条件下继续搅拌1h。过滤收集固体,加入到140ml 氢氧化钠溶液(2%),剧烈搅拌30min。过滤收集固体产物,用水洗涤后干燥。再用氯仿索氏提取,收集滤液,旋转蒸发得到对四氨基苯基卟啉。 对四硝基苯基钴卟啉(TNPPCo)的合成: 氮气保护下在装有冷凝管的100ml三口烧瓶中加入200mg T(P-NO 2)PPH 2 和 100mlDMF,加热至回流后在1h内分4次加入300mg(1.5mmol)CoCl 2·6H 2 O。继 续反应1h。溶液由紫色逐渐变成红黑色,TNPP在DMF里溶解度差,上金属后溶解度变大。蒸馏回收50mlDMF。然后用冰水冷却,加入50ml去离子水,过滤收集产物。用去离子水反复洗涤后用少量乙醇洗涤。干燥后得到0.2050g产物。
紫草宁生物合成途径中的代谢与调控 1.背景知识介绍 1.1 紫草及紫草宁 紫草(学名:Lithospermum erythrorhizon),为紫草科紫草属植物。又名山紫草、紫丹、紫草根,分布于日本、朝鲜以及中国大陆的辽宁、山西、湖南、甘肃、山东、湖北、广西、四川、陕西、贵州、江西、河北、河南等地,生长于海拔50米至2,500米的地区,多生长在山坡草地,目前尚未由人工引种栽培。紫草是一种重要的药用植物,其功效是凉血,活血,解毒透疹。用于血热毒盛,斑疹紫黑,麻疹不透,疮疡,湿疹,水火烫伤。紫草根部富含红色的萘醌类次生代谢产物——紫草宁及其衍生物。 紫草宁又称紫草素,英文名称:Shikalkin,英文别名: 5,8-Dihydroxy-2-(1-hydroxy-4-methylpent-3-enyl)naphthalene-1,4-dione,即5,8-二羟基-2-[(1R)-1-羟基-4-甲基戊-3-烯基]萘-1,4-二酮,结构式如下: 紫草宁为赤褐色针状晶体(由苯重结晶)。熔点149℃。旋光度-167°±10°(在苯中)。能溶于普通有机溶剂,以及甘油动植物油脂和碱性水溶液。难溶于碳酸氢碱溶液。与氢氧化碱金属作用显蓝色。 由于紫草素具有多种生物学活性,以紫草素为先导化合物开发抗炎、抗肿瘤、抗病毒新药的研究已成为热点课题,除此之外,紫草素还是良好的天然色素,已广泛用于食品、化妆品和印染工业中。 1.2紫草宁及其衍生物的药理作用
1.2.1 抗肿瘤活性 近年来,紫草次生代谢物的抗肿瘤活性倍受关注。紫草素能够抑制肝癌肿瘤细胞增殖[1]、诱导生殖系统肿瘤细胞凋亡[2],并兼具调控机体免疫的功能。紫草素在体外一定浓度范围内能抑制人白血病K562细胞增殖,诱导其凋亡。甲基丙烯酰紫草素具有较好的体内外抗肿瘤作用,作用机制可能与诱导细胞凋亡和抑制NF-zB p50的活性有关[3]。乙酰紫草素可通过诱导细胞凋亡来抑制胃癌SGC-7901细胞在体内外的增殖[4]。 1.2.2 抗炎活性 紫草素能有效减轻由中波紫外线(UVB)引起的表皮角蛋白细胞炎症,起到保护皮肤的作用;还可以减弱小神经胶质细胞的炎症反应,达到保护神经系统的作用。 1.2.3 降胆固醇活性 研究发现,从硬紫草根部氯仿提取物中分离出的三种化合物—乙酰紫草素、异丁基紫草素和β-羟基异戊酰紫草素均具有抑制人类酰基辅酶A-胆固醇酰基转移酶-1和人类酰基辅酶A-胆固醇酰基转移酶-2的活性。酰基辅酶A-胆固醇酰基转移酶是胆固醇生物合成途径的关键酶,乙酰紫草素、异丁基紫草素和β-羟基异戊酰紫草素通过抑制该酶的活性,从而达到降低胆固醇含量,防治动脉粥样化的目的。 紫草的药理作用除了上述内容之外,还有降血糖活性,抗生育、抗免疫缺陷、抗凝血、保肝护肝、抗前列腺素生物合成、抗菌及清除活性氧作用等。 1.3紫草及紫草宁的市场 紫草是我国传统中药材,多家中药饮片厂以紫草为主要原料研制开发生产了约500多种(规格)中成药、特药、新型中药,以及几十种中药饮片。这些产品投入市场后很受消费者欢迎,销量增加,对紫草的需求量也随之逐年大幅攀升。
项目名称:具有重要生物活性的天然产物的化学合 成 首席科学家:马大为中国科学院上海有机化学研究 所 起止年限:2010年1月-2014年8月 依托部门:上海市科委
一、研究内容 本项目的关键科学问题是针对具有重要生物活性的复杂天然产物,发展高效和实用的合成路线,以及阐明它们的结构–活性关系和作用机制。 本项目的将选择一批具有抗癌、抗炎、抗病毒和免疫等活性的生物碱、环酯肽、皂甙和萜类天然产物为研究对象,在综合运用化学各学科新概念、新知识和新技术的基础上,根据目标分子的结构进行巧妙设计,发展高效、高选择性的合成策略,实现一系列具有生物活性复杂天然产物的化学合成。在合成方式上将重点发展基于串联反应、多组分反应、无保护基合成、原子经济性、催化反应的应用和仿生合成等新合成策略。通过合成建立天然产物和其类似物的化合物库,与生物学家合作进行活性测试,总结相关天然产物的结构-活性关系。在此基础上发展用于化学生物学研究的天然产物分子探针,以发现相应天然产物的作用靶点。对于所发现的活性和选择性更好的化合物,我们将深入探索其成为治疗重要疾病药物的可能性。在进行目标分子的合成和相关生物学研究的同时,也将关注合成中的反应方法学问题,发展一些高效、选择性好、具有普适性的新合成方法。本项目具体的研究内容的如下: 1.开展一些具有具有抗癌、抗炎、抗菌、抗病毒等活性,结果新颖,目前还没 有全合成的报道,有一定的合成挑战性的天然产物进行全合成研究,争取实现它们的第一次全合成。这样的工作也为加快后续的构效关系研究和结构优化打下基础。所涉及的目标分子包括具有抗癌、抗炎、抗菌、抗病毒等活性的生物碱类化合物PF1270A/ B/C, Longeracinphyllin A, Sie b oldine A和Haouamine A/B;环肽类化合物Piperazimycin A, Chloptosin和Celogentin C;皂甙和萜类化合物Sepositoside A, Solanoeclepin A, Micrandilactone
2012.11.27-2010.12.10卟啉化合物的合成、理化性质及其应用 (苏州大学材料与化学化工学部09级化学类) 摘要:为了了解卟啉化合物,用郭灿城等人提出新方法合成TPPH2和CoTPP,并利用红外、紫外与荧光光谱分析其结构。 关键词:TPPH2、CoTPP、合成 Abstract:To understand the synthesis and token of Porphyrins,we synthetise TPPH2and CoTPP with new method proposed by Cancheng Guo et al,and characterized by FT-IR,UV and fluorescence spectrum. Keywords:TPPH2、CoTPP、synthetize 1.前言 卟啉(porphyrins)是卟吩(porphine)外环带有取代基的同系物和衍生物的总称,当其氮上2个质子被金属离子取代后即成金属卟啉配合物(metalloporphyrins)。自然界中存在许多天然卟啉及其金属配合物,如血红素、叶绿素、维生素B12、细胞色素P-450、过氧化氢酶等。天然卟啉化合物具有特殊的生理活性。人工合成卟啉来模拟天然卟啉化合物的各种性能一直是人们感兴趣和研究的重要课题。由于卟啉化合物独特的结构、优越的物理、化学及光学特征,使得卟啉化合物在仿生学、材料化学、药物化学、电化学、光物理与化学、分析化学、有机化学等领域都具有十分广阔的应用前景,正吸引着人们对卟啉化学不断深入地研究。 本实验采用郭灿城等人提出的合成四苯基卟啉的新方法,合成TPPH2和CoTPP,并利用红外、紫外与荧光光谱分析其结构。 2.实验部分 2.1、仪器与药品 仪器:烧杯(50mL×2、100mL×1)、量筒(50mL)、三颈烧瓶(250mL,19#×1/14#×2)、双颈烧瓶(50mL,19#×2)、茄形瓶(250mL,24#)、恒压滴液漏斗(14#)、球形冷凝管(19#)、干燥管(19#)、空心塞(19#×2、14#×2)、布氏漏斗及抽滤瓶、色谱柱(24#)、调压变压器、旋转蒸发仪、温度计(300℃)、油浴、磁力搅拌器、回流装置。
比拉斯汀的合成工艺研究 发表时间:2017-10-30T17:35:09.573Z 来源:《医药前沿》2017年10月第29期作者:徐连德1 徐琪琪2 徐英明2 [导读] 比拉斯汀(Bilastine),中文化学名为2-[4-(2-(4-(1-(2-乙氧基乙基)苯并咪唑-2-基)哌啶-1-基)乙基)苯基]-2-甲基丙酸。(1沂水县第一中学山东临沂 276405) (2山东罗欣药业集团股份有限公司山东临沂 276017) 【摘要】α,α-二甲基-4-(2-溴乙酰基)苯乙酸甲酯[2]经过还原反应制得α,α-二甲基-4-(2-溴乙基)苯乙酸甲酯[3],3与1-(2-乙氧基-乙基)-2-哌啶-4-基-1H-苯并咪唑[4]发生烷基化反应,再经水解得到比拉斯汀[1],总收率约76%。 【关键词】比拉斯汀;组胺H1受体拮抗剂;合成 【中图分类号】R976 【文献标识码】A 【文章编号】2095-1752(2017)29-0353-01 比拉斯汀(Bilastine),中文化学名为2-[4-(2-(4-(1-(2-乙氧基乙基)苯并咪唑-2-基)哌啶-1-基)乙基)苯基]-2-甲基丙酸,是西班牙FAES制药公司开发的第2代组胺H1受体拮抗剂,2012年欧盟批准其用于治疗变应性鼻炎及慢性特发性荨麻疹[1]。本品安全性良好,无常用抗组胺药物存在的镇静作用及心脏毒性,口服给药吸收迅速,具有良好的耐受性、安全性和较高的生物利用度[2]。 已有文献报道了1的合成路线[3-5]。本文选择以下路线α,α-二甲基-4-(2-溴乙酰基)苯乙酸甲酯[2]经过还原反应制得α,α-二甲基-4-(2-溴乙基)苯乙酸甲酯[3],3与1-(2-乙氧基-乙基)-2-哌啶-4-基-1H-苯并咪唑[4]发生烷基化反应,再经水解得到比拉斯汀[1],并进行了工艺优化。 文献[5]报道了由2制备3的过程,用三乙基硅烷-三氟乙酸进行还原,反应时间长达72h,收率91%。本研究通过调整三乙基硅烷-三氟乙酸的用量,控制回流反应温度,缩短了反应时间,收率90%。文献[5]由3制备1的过程中,3依次与2-(4-哌啶基)-1H-苯并咪唑和2-氯乙基乙醚发生亲核取代反应后,水解得比拉斯汀,反应步骤长,且操作繁琐,且3在与2-(4-哌啶基)-1H-苯并咪唑发生亲核取代反应时,咪唑环上的氮-氢不可避免的会与哌啶基上的氮-氢进行竞争,生成副产物,影响收率和纯度。本研究在文献基础上进行了改进,将3直接与4进行烷基化反应,再进行水解,一锅法制备1,方法工艺简单,操作简便,收率及产品纯度均有较大幅度提高,总收率为76%,适合工业化生产。 图1 1的合成路线 Fig.1 Synthetic Route of 1 1.实验部分 1.1 α,α-二甲基-4-(2-溴乙基)苯乙酸甲酯[3]的制备 冰浴冷却下分别向反应瓶内加入20mL二氯甲烷、α,α-二甲基-4-(2-溴乙酰基)苯乙酸甲酯(30.0g,100.7mmol,采用文献方法[5]制得,纯度99.1%)、三氟乙酸(36mL,484.6mmol)、三乙基硅烷(18mL,112.7mmol)。冰浴下搅拌30min后,升温至60℃回流反应20h。反应结束,滴加饱和碳酸溶液(约150ml),加入乙酸乙酯(100ml*2)萃取,有机相浓缩干燥,得无色油状物(25.7g,90%)(文献:91%[5])。ESI-MS,m/z(%):307[M+Na]+,283[M-H]+。元素分析:C13H17BrO2,实测值(计算值)%:C54.96(55.00);H6.02(6.04);Br28.11(28.14);O11.27(11.27)。 1.2 比拉斯汀[1]的合成 在反应瓶中加入3(99.36g,0.35mol)和4(82.01g,0.3mol,购自:江苏弘和药物研发有限公司,纯度98%),搅拌下加入10ml聚乙二醇-400和45ml水,在冰水浴的冷却下慢慢加入混合碱(0.25molNaOH+0.1molNa2CO3),于40℃下快速搅拌3.5小时后放置,使反应液冷却至室温,加入3N丁二酸溶液2.1L,加热回流24小时,用10%氢氧化钠水溶液调至pH=7,用乙醚(450ml*2)萃取,旋出溶剂,得到固体1(116.83g,84%),mp291~293℃(文献:295-296[5])。纯度为99.8% [HPLC归一化法:同文献[5]。ESI-MS,m/z(%):487[M+Na]+,463[M-H]+。元素分析:C28H37N3O3,实测值(计算值)%:C72.50(72.54);H8.01(8.04);N9.07(9.06); O10.38(10.35)。 【参考文献】 [1] Corc6stegui R,Labeaga L,Inneririty A,et a1.Preclinical pharmacology of bilastine,a new selective histamine Hl receptor antagonist:receptor selectivity and in vitro anti-histaminic activity[J].Drugs R D,2005,6(6):371-384. [2] Carter NJ.Bilastine:in allergic rhinitis and urticaria [J].Drugs,2012,72(9):1257-1269. [3] Lee CH,Khoo JH,Kwon KC,eta1.Process for preparation of 2-methyl-2-phenylpropionic acid derivatives and novel intermediate compounds:WO,2009102155[P].2009-02-12. [4]王蕾,李科,王倩,等.2-(4-卤乙基)苯基-2-甲基丙酸酯的制备方法及合成比拉斯汀的方法:中国,102675101[P].2012-09-19. [5]孔昊,耿海明,梅玉丹,等.比拉斯汀的合成[J].中国医药工业杂志,2015,46(7):677-679.