实验六实时定量PCR技术
实时荧光定量PCR技术于1996年由美国Applied Biosystems公司推出,由于该技术不仅实现了PCR从定性到定量的飞跃,而且与常规PCR相比,它具有特异性更强、有效解决PCR污染问题、自动化程度高等特点,目前已得到广泛应用。实时荧光定量PCR仪包括热循环仪,检测系统,计算机及软件分析系统。
一、实验原理
谓实时荧光定量PCR技术,是指在PCR反应体系中加入荧光基团,利用荧光信号积累实时监测整个PCR进程,最后通过标准曲线对未知模板进行定量分析的方法。
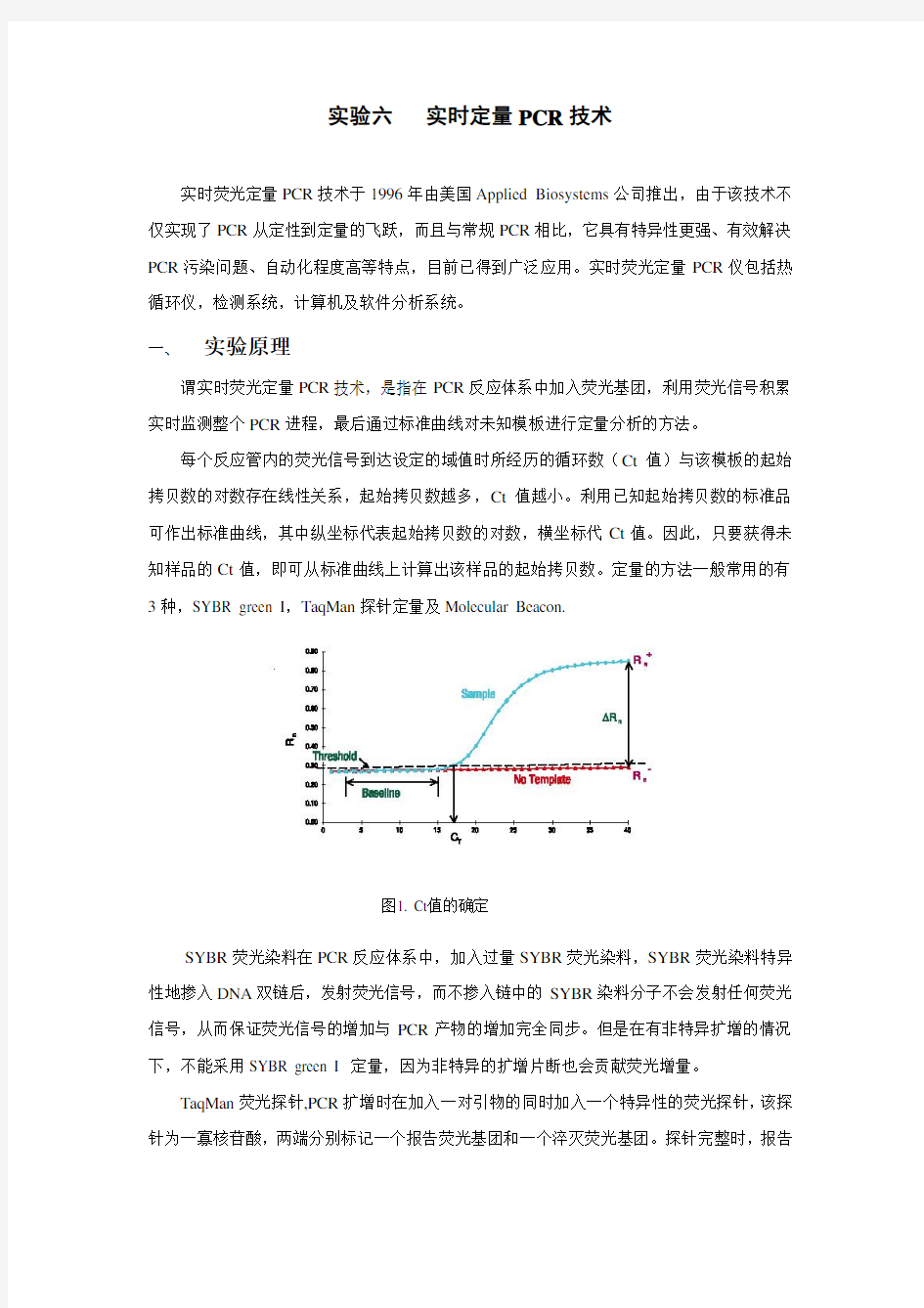
每个反应管内的荧光信号到达设定的域值时所经历的循环数(Ct值)与该模板的起始拷贝数的对数存在线性关系,起始拷贝数越多,Ct值越小。利用已知起始拷贝数的标准品可作出标准曲线,其中纵坐标代表起始拷贝数的对数,横坐标代Ct值。因此,只要获得未知样品的Ct值,即可从标准曲线上计算出该样品的起始拷贝数。定量的方法一般常用的有3种,SYBR green I,TaqMan探针定量及Molecular Beacon.
图1. Ct值的确定
SYBR荧光染料在PCR反应体系中,加入过量SYBR荧光染料,SYBR荧光染料特异性地掺入DNA双链后,发射荧光信号,而不掺入链中的SYBR染料分子不会发射任何荧光信号,从而保证荧光信号的增加与PCR产物的增加完全同步。但是在有非特异扩增的情况下,不能采用SYBR green I 定量,因为非特异的扩增片断也会贡献荧光增量。
TaqMan荧光探针,PCR扩增时在加入一对引物的同时加入一个特异性的荧光探针,该探针为一寡核苷酸,两端分别标记一个报告荧光基团和一个淬灭荧光基团。探针完整时,报告
基团发射的荧光信号被淬灭基团吸收;PCR扩增时,Taq酶的5’-3’外切酶活性将探针酶切降解,使报告荧光基团和淬灭荧光基团分离,从而荧光监测系统可接收到荧光信号,即每扩增一条DNA链,就有一个荧光分子形成,实现了荧光信号的累积与PCR产物形成完全同步. TaqMan探针特异性地结合在靶序列上,只有真正发生靶序列扩增的反应才会有荧光增量的产生。
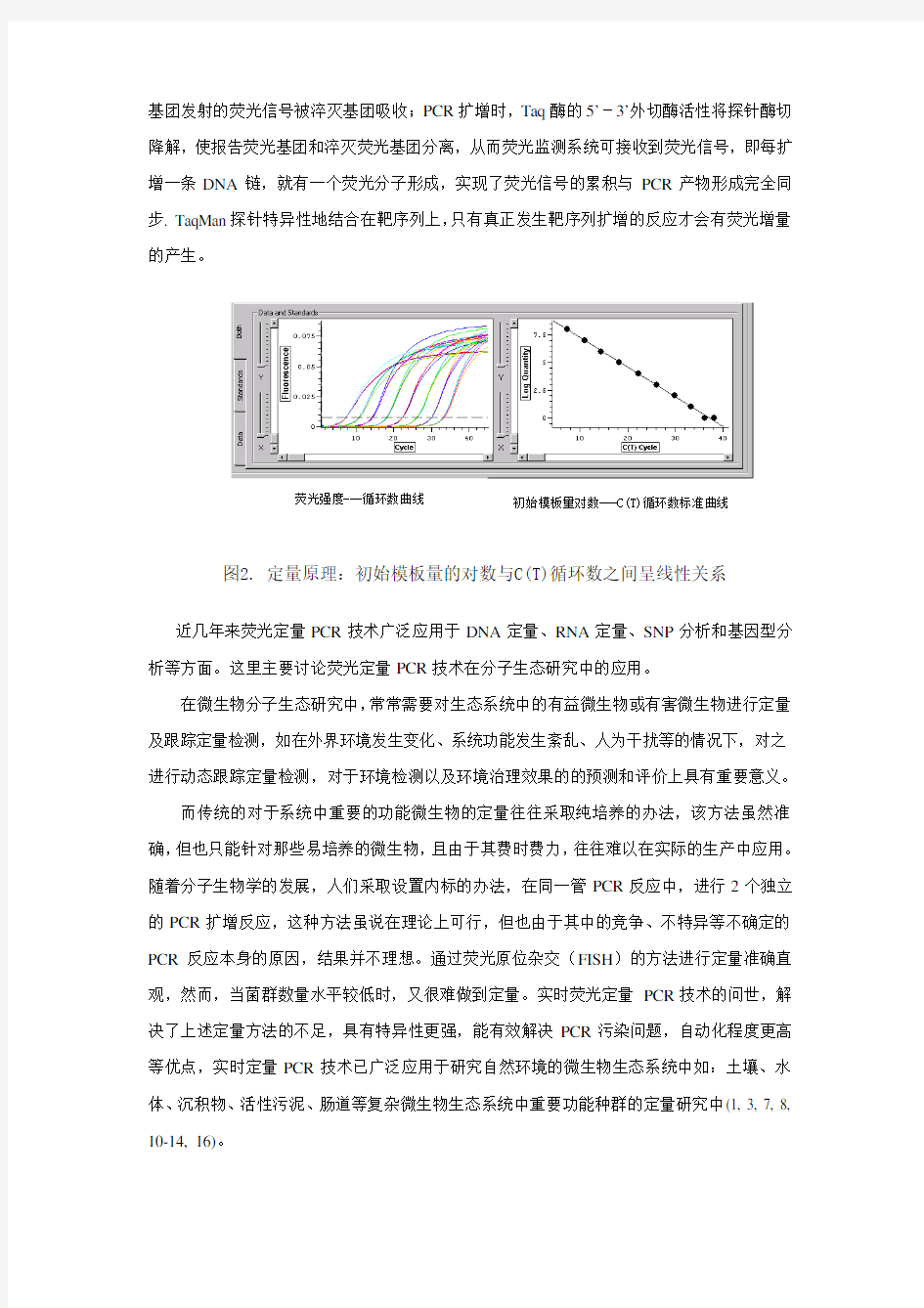
荧光强度---循环数曲线初始模板量对数---C(T)循环数标准曲线
图2. 定量原理:初始模板量的对数与C(T)循环数之间呈线性关系
近几年来荧光定量PCR技术广泛应用于DNA定量、RNA定量、SNP分析和基因型分析等方面。这里主要讨论荧光定量PCR技术在分子生态研究中的应用。
在微生物分子生态研究中,常常需要对生态系统中的有益微生物或有害微生物进行定量及跟踪定量检测,如在外界环境发生变化、系统功能发生紊乱、人为干扰等的情况下,对之进行动态跟踪定量检测,对于环境检测以及环境治理效果的的预测和评价上具有重要意义。
而传统的对于系统中重要的功能微生物的定量往往采取纯培养的办法,该方法虽然准确,但也只能针对那些易培养的微生物,且由于其费时费力,往往难以在实际的生产中应用。随着分子生物学的发展,人们采取设置内标的办法,在同一管PCR反应中,进行2个独立的PCR扩增反应,这种方法虽说在理论上可行,但也由于其中的竞争、不特异等不确定的PCR反应本身的原因,结果并不理想。通过荧光原位杂交(FISH)的方法进行定量准确直观,然而,当菌群数量水平较低时,又很难做到定量。实时荧光定量 PCR技术的问世,解决了上述定量方法的不足,具有特异性更强,能有效解决PCR污染问题,自动化程度更高等优点,实时定量PCR技术已广泛应用于研究自然环境的微生物生态系统中如:土壤、水体、沉积物、活性污泥、肠道等复杂微生物生态系统中重要功能种群的定量研究中(1, 3, 7, 8, 10-14, 16)。
例如,在对人的肠道微生物生态学的研究中,对肠道中各类微生物的定量研究有很多文献报道。如对肠道中起重要作用的一个功能菌——双歧杆菌(Bifidobacteria)的定量。健康成人肠道中大约含有1014个细菌细胞,这个数量大约是人体器官细胞数的10倍,如此庞大的微生物量,对人体的健康起着及其重要的作用,而不同个体又有自身所特有的微生物菌群结构。肠道功能的紊乱往往与肠道微生物菌群结构的变化(失调)紧密相关,那么对肠道菌群的动态变化进行跟踪研究就显得尤为重要。双歧杆菌是儿童和成人肠道中的重要的优势功能菌之一,可以降解人体不易消化的碳水化合物,可治疗便秘,慢性腹泻,从而保护肝脏,防治高血压和动脉硬化,改善乳糖消化不良症,起营养作用和抗衰老作用。而儿童和成人体内的数量却有所不同,对这一重要功能菌的定量以及定量地跟踪该菌在不同健康状态下的动态变化具有重要意义。设计双歧杆菌的专一性(genus- or species-specific)的引物,提取肠道微生物混合的总的基因组DNA,利用SYBR green I 或Taqman探针进行实时的荧光定量,就可以很快速便捷的对一系列动态跟踪的样品进行定量,也可以大规模的平行比较其数量的多少(10)。Favier等人对不同年龄的人体肠道中乳酸杆菌的量进行了检测(9)。Bartosch等人对健康老年人和住院老年人,以及抗生素治疗对老年人肠道微生物菌群结构的影响进行了定量检测,发现住院老年人中菌群16S rDNA的拷贝数明显低于健康老年人的,抗生素治疗对菌群结构影响也较大(2)。Favier等人用该技术结合DGGE连续10个月检测了健康婴儿肠道微生物的演替过程(6)。
乳酸杆菌产生的有机酸被认为是引起牙周脱钙的主要原因,从而引起牙齿脱落,Byun et al. 等人对65例牙龋病人口腔中的乳酸菌进行了定量研究 (4),发现该类菌确实大量存在于牙龋病人的口腔中。实时定量PCR技术也被用来分析食草动物食性改变后瘤胃内优势微生物种群的动态变化 (17)。
Pseudoalteromonas species是海洋中的一个非常重要的微生物,与多种海洋真核生物相关,其所产生的活性物质具有抗细菌、除海藻、抗真菌等作用,Skovhus 等人用实时定量的PCR技术对海洋样品中该种群的数量进行了评估 (15)。Dionisi等人将该技术应用于研究活性污泥中,MLVSS 值在260到2,610 mg/L时,Nitrospira数量的多少(5)。应用实时定量PCR 技术研究自然界复杂微生物群落中重要功能菌的数量,以及外界条件发生变化时其数量动态变化的检测,诸如此类的研究报道很多,在这里我们以定量猪肠道内拟杆菌的数量为例,介绍实时荧光定量PCR技术的使用方法。
二、实验目的
定量待测样品中靶DNA含量的多少及其动态变化。
三、实验材料
(1)试剂:
Ex Taq酶系统(TaKaRa)
标准质粒:拟杆菌16S rDNA特异片断的克隆质粒
仔猪肠道微生物总的基因组DNA
10 × SYBR green 1 (Amerson)
dNTP
引物:拟杆菌特异(group-specific)引物
无菌双蒸水
(2)仪器设备
MJ optical 2 real-time 定量PCR仪
PCR八联管
四、操作步骤
1.10倍拷贝数梯度稀释标准的DNA样品(如108,107,106,105),用于制作标准曲
线(标准样品一般为经限制性内切酶酶切后的线性质粒);
2.准备好待测DNA样品;
3.调配反应液,可能不同的扩增反应需要的扩增条件如Mg2+浓度,引物浓度等不同,
需要逐一摸索,这里提供一个一般的条件,
试剂加量(μl) ×10
148.5 ddH2O 14.85
20
dNTP 2
10×Buffer 2.5 25
Primer 1 0.5 5
Primer 2 0.5 5
Template 2 20
Taq ase 0.15 1.5
10×SYBR green 1 2.5 25
(23μl 分装)
230
∑ 25
4.分装后逐一加入模板,设置阴性对照,设至少3个标准样品,用于生成标准曲线;
设置1管阴性对照,其它为待测样品;
5.放入到定量PCR仪中,设置样品号,设置程序,运行程序;
6.实时查看扩增情况;
7.运行结束后,生成标准曲线,记录样品的拷贝数,用于数据分析;
8.还可以测定产物的融解曲线,计算其Tm值。
五、注意事项
1.标准样品最好为待测目标DNA克隆后的质粒,经酶切为线性DNA, 需要过柱纯化,测定浓度,计算拷贝数,制备好稀释梯度的DNA样品,稀释的好坏直接影响标准曲线的质量,也就直接影响的定量结果的可靠性;
2. 操作过程要十分小心,避免分装到八联管时发生污染;
3.设定阴性对照。
六、参考文献
1. Bach, H.-J., J. Tomanova, M. Schloter, and J. C. Munch. 200
2. Enumeration of total bacteria
and bacteria with genes for proteolytic activity in pure cultures and in environmental samples
by quantitative PCR mediated amplification. Journal of Microbiological Methods 49:235-245.
2. Bartosch, S., A. Fite, G. T. Macfarlane, and M. E. T. McMurdo. 2004. Characterization of
Bacterial Communities in Feces from Healthy Elderly V olunteers and Hospitalized Elderly
Patients by Using Real-Time PCR and Effects of Antibiotic Treatment on the Fecal Microbiota.
Appl. Environ. Microbiol. 70:3575-3581.
3. Bowers, H. A., T. Tengs, H. B. Glasgow, Jr., J. M. Burkholder, P. A. Rublee, and D. W. Oldach.
2000. Development of Real-Time PCR Assays for Rapid Detection of Pfiesteria piscicida and Related Dinoflagellates. Appl. Environ. Microbiol. 66:4641-4648.
4. Byun, R., M. A. Nadkarni, K. L. Chhour, F. E. Martin, N. A. Jacques, and N. Hunter. 2004.
Quantitative analysis of diverse lactobacillus species present in advanced dental caries. J Clin Microbiol 42:3128-36.
5. Dionisi, H. M., G. Harms, A. C. Layton, I. R. Gregory, J. Parker, S. A. Hawkins, K. G.
Robinson, and G. S. Sayler. 2003. Power analysis for real-time PCR quantification of genes
in activated sludge and analysis of the variability introduced by DNA extraction. Appl Environ Microbiol 69:6597-604.
6. Favier, C. F., E. E. Vaughan, W. M. De Vos, and A. D. L. Akkermans. 2002. Molecular
Monitoring of Succession of Bacterial Communities in Human Neonates. Appl. Environ.
Microbiol. 68:219-226.
7. Gerard, C., K. Olsson, R. Ramanathan, C. Reading, and E. Hanania. 1998. Improved
quantitation of minimal residual disease in multiple myeloma using real-time polymerase
chain reaction and plasmid-DNA complementarity determining region III standards. Cancer
Res 58:3957-3964.
8. Gruntzig, V., S. C. Nold, J. Zhou, and J. M. Tiedje. 2001. Pseudomonas stutzeri Nitrite
Reductase Gene Abundance in Environmental Samples Measured by Real-Time PCR. Appl.
Environ. Microbiol. 67:760-768.
9. Guan, L. L., K. E. Hagen, G. W. Tannock, D. R. Korver, G. M. Fasenko, and G. E. Allison.
2003. Detection and Identification of Lactobacillus Species in Crops of Broilers of Different
Ages by Using PCR-Denaturing Gradient Gel Electrophoresis and Amplified Ribosomal DNA Restriction Analysis. Appl. Environ. Microbiol. 69:6750-6757.
10. Gueimonde, M., S. Tolkko, T. Korpimaki, and S. Salminen. 2004. New Real-Time
Quantitative PCR Procedure for Quantification of Bifidobacteria in Human Fecal Samples.
Appl. Environ. Microbiol. 70:4165-4169.
11. Harms, G., A. Layton, H. Dionisi, V. Garrett, S. Hawkins, K. Robinson, and G. Sayler. 2003.
Real-time PCR quantification of nitrifying bacteria in a municipal wastewater treatment plant.
Environ Sci Technol 37:343-351.
12. Hermansson, A., and P.-E. Lindgren. 2001. Quantification of Ammonia-Oxidizing Bacteria in
Arable Soil by Real-Time PCR. Appl. Environ. Microbiol. 67:972-976.
13. Hristova, K. R., C. M. Lutenegger, and K. M. Scow. 2001. Detection and Quantification of
Methyl tert-Butyl Ether-Degrading Strain PM1 by Real-Time TaqMan PCR. Appl. Environ.
Microbiol. 67:5154-5160.
14. Skovhus, T. L., N. B. Ramsing, C. Holmstrom, S. Kjelleberg, and I. Dahllof. 2004. Real-Time
Quantitative PCR for Assessment of Abundance of Pseudoalteromonas Species in Marine
Samples. Appl. Environ. Microbiol. 70:2373-2382.
15. Skovhus, T. L., N. B. Ramsing, C. Holmstrom, S. Kjelleberg, and I. Dahllof. 2004. Real-time
quantitative PCR for assessment of abundance of Pseudoalteromonas species in marine
samples. Appl Environ Microbiol 70:2373-82.
16. Stults, J. R., O. Snoeyenbos-West, B. Methe, D. R. Lovley, and D. P. Chandler. 2001.
Application of the 5' Fluorogenic Exonuclease Assay (TaqMan) for Quantitative Ribosomal
DNA and rRNA Analysis in Sediments. Appl. Environ. Microbiol. 67:2781-2789.
17. Tajima, K., R. I. Aminov, T. Nagamine, H. Matsui, M. Nakamura, and Y. Benno. 2001.
Diet-dependent shifts in the bacterial population of the rumen revealed with real-time PCR.
Appl Environ Microbiol 67:2766-74.
(本章作者:魏桂芳)
实时定量PCR技术原理 提要 □基本概念 □定量PCR的数学原理 □定量PCR的化学原理 □等位基因鉴定原理 □定量PCR仪光学原理 基本概念 什么是PCR? ?PCR能否只用一条引物?三条引物? ?PCR能否使用ddNTP? 典型的PCR四阶段 PCR理论方程 N = N ×(1+e)n N:产物分子数 :起始分子数 N e:扩增效率 n:循环次数 循环次数
起点定量与终点定量 起点量是样本中原来的DNA量,更有意义; 终点量经过PCR 放大,并非研究所期望的数据□ 起点定量误差小,终点定量误差大 标准曲线方法 通过CT值测定起始DNA量 定量PCR的数学原理 值荧光信号有统计学意义显著增长(即穿过阈值线)时的循环次数 什么是C T 从基线到指数增长的拐点所对应的循环次数 线性图谱
C 值 T 半对数图谱 C 值 T ?浓度增加1倍,CT值减小1个单位 ?浓度增加10倍,CT值减小3.3个单位 为什么C ∝起始DNA浓度? T 值与起始DNA浓度的关系 C T ?单拷贝检测在理论和实验上能做到吗?
?最理想的标准曲线斜率是多少? 什么是阈值 基线信号的标准偏差×10 基线→阈值→C 值 T 基线调整规则 值>18,不需调整,使用自动分析结果 如果C T 值<18,修改终点,再分析一次 如果C T 起点:进口试剂取3,国产试剂取6 终点:最小的CT值–4,通常为15 长度:大于或等于6个循环 PCR方程只在指数期成立 CT值和阈值必须位于指数增长阶段内
手工数据分析方法 1.实验结束,软件根据默认值的基线(3-15)自动分析,给出结果和图谱 2.如果C T 值> 18,使用自动分析结果,出报告 3.如果有C T 值<18,根据[最小的C T 值-4] 修改基线终点,重新分析数据 软件自动的数据分析方法 软件自动计算全部参数:基线、阈值、CT值、浓度 ?能否用鼠标拖阈值线以正确区分阴性与阳性? ?阈值与DNA浓度成反比,对吗?
实时荧光定量PCR原理和实验 陈云地 作者单位:200030 美国应用生物系统公司(Applied Biosystems) 无论是对遗传病(如地中海贫血和血友病)、传染病(如肝炎和艾滋病)或肿瘤进行基因诊断,还是研究药物对基因表达水平的影响,或者监控药物和疗法的治疗效果,定量PCR技术都可以发挥很大作用。定量PCR技术的最新进展是实时荧光定量。该技术借助于荧光信号来检测PCR产物,一方面提高了灵敏度,另一方面还可以做到PCR每循环一次就收集一个数据,建立实时扩增曲线,准确地确定CT值,从而根据CT值确定起始DNA拷贝数,做到了真正意义上的DNA定量。这是DNA定量技术的一次飞跃。 根据最终得到的数据不同,定量PCR可以分为相对定量和绝对定量两种。典型的相对定量如比较经过不同方式处理的两个样本中基因表达水平的高低变化,得到的结果是百分比;绝对定量则需要使用标准曲线确定样本中基因的拷贝数或浓度。根据所使用的技术不同,荧光定量PCR 又可以分为TaqMan探针和SYBR Green I 荧光染料两种方法。比较而言,探针杂交技术在原理上更为严格,所得数据更为精确;荧光染料技术则成本更为低廉,实验设计更
为简便。在选择实验方案时要根据实验目的和对数据精度的要求来决定。 定量实验与定性实验最大的不同,是要考虑统计学要求并对数据进行严格的校正,以消除偶然误差。因此重复实验和设立内对照非常重要。由于各种各样的客观原因,这一点在实践中往往被轻视或忽视,需要着重强调。当然,与定性实验一样,定量PCR也要设立阴性和阳性对照,以监控试剂和实验操作方面可能出现的问题。 1 为什么终点定量不准确? 我们都知道理论上PCR是一个指数增长的过程,但是实际的PCR扩增曲线并不是标准的指数曲线,而是S形曲线。这是因为随着PCR循环的增多,扩增规模迅速增大,Taq酶、dNTP、引物,甚至DNA模板等各种PCR要素逐渐不敷需求,PCR的效率越来越低,产物增长的速度就逐渐减缓。当所有的Taq酶都被饱和以后,PCR就进入了平台期。由于各种环境因素的复杂相互作用,不同的PCR反应体系进入平台期的时机和平台期的高低都有很大变化,难以精确控制。所以,即使是重复实验,各种条件基本一致,最后得到的DNA拷贝数也是完全不一样的,波动很大(图1)。
实时荧光定量PCR(Real-Time PCR)实验流程 一、RNA的提取(详见RNA提取及反转录) 不同组织样本的RNA提取适用不同的提取方法,因为Real-Time PCR对RNA样品的质量要求较高,所以,正式实验前要选择一款适合自己样品的提取方法,在实验过程中要防止RNA的降解,保持RNA的完整性。 在总RNA的提取过程中,注意避免mRNA的断裂;取2ug进行RNA的甲醛变性胶电泳检测,如果存在DNA污染时,要用DNase I进行消化(因为在处理过程中RNA极易降解,建议体系中加入适量RNA酶抑制剂)。 二、DNase I 消化样品RNA 中的DNA 用DNase I 消化DNA 组份加量 模板(RNA) 10ug RNase Inhibitor 4ul DNase I buffer 10ul DNase I 10ul DEPC处理H2O 至100ul 混匀,37℃ 90min 三、RNA琼脂糖凝胶电泳 1.1%的琼脂糖凝胶电泳凝胶的配制: 1)称取琼脂糖0.45g放入三角瓶中,向其中加入4.5ml的10×MOPS缓冲液和39.5ml 的DEPC水,放微波炉里溶化。 2)待冷却到60摄氏度左右时,加入1ml甲醛,摇匀(避免产生气泡)。倒入凝胶板上凝固30min。 2.取各个RNA样品4μl,加入6×RNA电泳上样缓冲液2μl混匀,加入变性胶加样孔中。3.120V电压下电泳25min。用凝胶紫外分析仪观察,照相保存。 4.RNA电泳结果如下图所示。可见28S和18S两条明亮条带,无DNA条带污染。 四.RNA反转录为cDNA 反转录程序(以MBI的M-MLV为例) 组份加量(20ul体系) 加量(40ul体系) 模板(RNA) 0.1~2.5ug(根据条带的亮度适当调整) 3ug(根据条带的亮度适当调整) 引物T18(50uM)(或其他引物) 2.0ul 4.0ul DEPC处理H2O 至12.5ul 至25ul
实时荧光定量PCR方法简介 一.实时荧光定量PCR的基本原理 理论上,PCR过程是按照2n(n代表PCR循环的次数)指数的方式进行模板的扩增。但在实际的PCR反应过程中,随着反应的进行由于体系中各成分的消耗(主要是由于聚合酶活力的衰减)使得靶序列并非按指数方式扩增,而是按线性的方式增长进入平台期。因此在起始模板量与终点的荧光信号强度间没有可靠的相关性。如采用常规的终点检测法(利用EB染色来判断扩增产物的多少,从而间接的判断起始拷贝量),即使起始模板量相同经PCR 扩增、EB染色后也完全有可能得到不同的终点荧光信号强度。 为了能准确判断样品中某基因转录产物(mRNA)的起始拷贝数,实时荧光定量PCR采用新的参数——Ct值,定量的根本原理是Ct值与样品中起始模板的拷贝数的对数成线性反比关系。 Ct值是如何得到的 在实时荧光定量PCR的过程中,靶序列的扩增与荧光信号的检测同时进行,定量PCR仪全程采集荧光信号,实验结束后分析软件自动按数学算法扣除荧光本底信号并设定阈值从而得到每个样品的Ct值。 Ct值的定义 Ct值中的“C”代表Cycle(循环),“t”代表检测threshhold(阈值),其含义是PCR扩增过程中荧光信号强度达到阈值所需要的循环数;也可以理解为扩增曲线与阈值线交点所对
应的横坐标。 Ct值与样品中模板的对应关系 Ct值与样品中起始模板的拷贝数的对数成线性反比关系(y=ax+b,x代表起始模板拷贝数的对数,y代表Ct值)。 与终点法相比利用Ct值的优势 由于Ct值是反映实际PCR反应过程中扩增即将进入指数期的参数,该参数几乎不受试剂消耗等因素的影响,因此利用Ct值判断的起始模板拷贝数更加精确,重复性也更好。传统的终点检测法是在PCR扩增经历了指数扩增期进入平台期后利用EB等染料染色来判断扩增产物的多少,从而间接的判断起始拷贝量,这种方法的精确度不高、重复性也不好。 下图中是96个复孔的实时扩增曲线(完全相同的反应体系、相同的反应protocol、相同的样品起始浓度),可以看到Ct值具有很好的重复性,而终点的荧光信号强度差异达到300个单位。 此外,采用实时荧光定量PCR还能从方法学上有效的防止PCR实验中交叉污染的问题。因为荧光定量PCR中模板的扩增与检测是同时进行的,当实验完成后即可获得定量结果,
实时定量PCR技术综述 一.基本原理 1.PCR反应动力学 右图为PCR反应曲线(横坐标为循环数,纵坐标表征产物量),不同的曲线代表初始模板量不同的 PCR反应。PCR反应的动力学公式为: C n=C0(1+E)n 其中C0和C n分别为初始模板和n循环的拷贝数,n为循环数,E为扩增效率(0≤E≤1),理想状态下(即每个循环中所有模板均与引物结合并等到扩增),E为1。 由上图可知,只有在线性区段E值才为定值,这样才能通过检测C n定量C0,由于终点检测不能保证在线性区段,所以重复性不好,实时检测(每个循环检测一次)技术解决了这个问题。 2.定量PCR原理 定量PCR是将待测标本与一系 列浓度(C0)已知的标准品在同 一条件下共同扩增,并进行实时 检测,根据标准品的检测值及已 知的C0值作出标准曲线,如右
图(横作标为的C0对数值),再根据待测标本的检测值在标准曲线上查到待测标本的C0值。 3.信号产生 目前定量PCR技术信号产生都是基于FRET (fluorescence resonance energy transfer)的原理,即在一定波长的光激发下,一个荧光基团A发光,并且被邻近的另一个荧光基团B 吸收,使荧光基团B发光。如检测荧光基团A,应在FRET消除后;如检测B,则应在FRET 发生时检测。 根据信号产生方式的不同可将定量PCR技术分为三类:TaqMan Probes技术;Hybridization Probes技术;Molecular Beacons技术。 TaqMan Probes 技术(右图A)是 Perkin Elmer公司 1995年研制,利用 热稳定DNA聚合 酶5’→3’外切酶活 性,将TaqMan 探 针5’端荧光基团切 去,使之与3’端荧 光基团分离、荧光 淬灭消失,在特定 波长下检测5’端荧 光基团即可表征 PCR产物的量。 Hybridization Pro bes技术(右图B)是Roche公司建立的,PCR反应退火过程中,两个探针与模板杂交(相邻一个碱基),发生FRET,这时检测第二个荧光基团即可表征PCR产物的量。Hybridization Probes技术要求热稳定DNA聚合酶不具备5’→3’外切酶活性,而是在引物延伸过程中被取代,使探针可在下一循环再与模板杂交,如探针被降解,可导致定量不准。
实时荧光定量PCR仪ViiA 7 操作步骤 ——以RNase P示例实验为例 一、定义384孔样品模块的实验属性 打开电脑访问ViiA 7 软件,然后打开左侧仪器开关。单击Experiment Setup图标。单击Experiment Properties以访问Experiment Properties屏幕。 在ViiA 7 软件中设计RNase P实验示例时,请输入: 二、使用Define屏幕定义RNase P示例实验的目标基因、样品。 1. 单击Define以访问Define屏幕。 2. 定义目标基因 a. 单击New以增加和定义目标基因。 b. 在目标基因表中,单击Target Name列中的一个单元格,并输入: c. (可选)单击Save以便将新增或原有的正在编辑的目标基因保存到Target Library。 d. 单击Add Saved从目标基因库添加目标基因。 3. 定义样品 a. 单击New以增加和命名样品。 b. 在样品表中,单击Sample Name列中的一个单元格,并输入: c. (可选)单击Save以将新增或原有的正在编辑的样品保存到Sample Library。 d. 单击Add Saved从样品库添加样品。 4. (可选)定义生物学平行测定 a. 在Define Biological Replicates Groups表中,单击New以增加和命名生物学平行 测定组。 b. 从下拉菜单选择Color。 c. 单击Comments列,以便为该生物学平行测定组添加注释。 注:实验示例不使用生物学平行测定组。保留Biological Replicate Groups空白。 5. 选择用作参比荧光的染料ROX。
实时荧光定量 PCR 技术简介 实时荧光定量PCR(Quantitative Real-time PCR)是一项以PCR 反应为基础的DNA 定量技术,通过对目标基因在扩增过程中产生的拷贝数进行实时的定量,从而达到对目的基因 的定性和定量分析。现有两种常用的方法对PCR 产物进行荧光定量:一种是利用荧光染料 与双链DNA 结合,通过荧光强度进行定量;另一种是利用携带了荧光报告基团的特异DNA 探针对目标基因进行定量。 一、利用荧光染料进行定量 一种最为常用的定量方法就是在PCR 反应体系中加入荧光染料,此类荧光染料会与所 有的双链DNA 结合,并产生荧光。游离的荧光分子不会产生荧光信号,只有与双链DNA 结合的荧光分子才会释放荧光,随着DNA 拷贝数的增加,测得的荧光强度也会增强。 利用荧光染料进行定量的优势就是成本低廉,只需要一对普通的引物就能完成定量。然而,常用的诸如SYBR Green 染料会与所有的双链DNA 无差别地结合,包括引物二聚体,因 此有可能会导致对目标基因的定量不精确,灵敏度偏低。 二、利用荧光探针进行定量 荧光探针只能检测出与自身序列互补的DNA 片段,因此用探针法定量可以有效地避免引物二聚体的干扰,使定量结果更加精确。此外,通过使用携带不同荧光信号的多种探针,我 们可以同时对一个样品中的多个靶序列进行定量。 荧光探针的5’端携带有一个荧光报告基团,3’端则为淬灭基团,在正常情况下两个 基团间的距离很近,淬灭基团会抑制报告基团使其无法发出荧光。在PCR 反应过程中,引 物和荧光探针在退火阶段都会与目的片段结合;在延伸阶段,Taq 酶因为具有5’-3’核酸 外切酶活性,会将探针,使得报告基团和淬灭基团相互分开,从而释放出荧光。每增加一条目 的基因的拷贝,就会有一个探针被切开并释放荧光信号,因此随着PCR 反应的进行,荧光 信号会逐渐增强。 1 / 2
实时荧光定量PCR全方位解析 实时荧光定量PCR技术于1996年由美国Applied Biosystems公司推出,所谓实时荧光定量PCR技术,是指在PCR反应体系中加入荧光基团,利用荧光信号积累实时监测整个PCR进程,最后通过标准曲线对未知模板进行定量分析的方法。 PCR原理 所谓实时荧光定量PCR技术,是指在PCR反应体系中加入荧光基团,利用荧光信号积累实时监测整个PCR进程,最后通过标准曲线对未知模板进行定量分析的方法。 1. Ct 值的定义 在荧光定量PCR技术中,有一个很重要的概念-- Ct值。C代表Cycle,t代表threshold,Ct值的含义是:每个反应管内的荧光信号到达设定的域值时所经历的循环数。 2. 荧光域值(threshold)的设定 PCR反应的前15个循环的荧光信号作为荧光本底信号,荧光域值的缺省设置是3-15个循环的荧光信号的标准偏差的10倍,即:threshold = 10 ′ SDcycle 6-15 3. Ct值与起始模板的关系 研究表明,每个模板的Ct值与该模板的起始拷贝数的对数存在线性关系〔1〕,起始拷贝数越多,Ct值越小。利用已知起始拷贝数的标准品可作出标准曲线,其中横坐标代表起始拷贝数的对数,纵坐标代Ct值。因此,只要获得未知样品的Ct值,即可从标准曲线上计算出该样品的起始拷贝数。 4. 荧光化学 荧光定量PCR所使用的荧光化学可分为两种:荧光探针和荧光染料。现将其原理简述如下: 1)TaqMan荧光探针:PCR扩增时在加入一对引物的同时加入一个特异性的荧光探针,该探针为一寡核苷酸,两端分别标记一个报告荧光基团和一个淬灭荧光基团。探针完整时,报告基团发射的荧光信号被淬灭基团吸收;PCR扩增时,Taq酶的5'-3'外切酶活性将探针酶切降解,使报告荧光基团和淬灭荧光基团分离,从而荧光监测系统可接收到荧光信号,即每扩增一条DNA链,就有一个荧
实时荧光定量PCR技术的原理及应用(图) 一、实时荧光定量PCR原理 (一)定义:在PCR反应体系中加入荧光基团,利用荧光信号累积实时监测整个PCR进程,最后通过标准曲线对未知模板进行定量分析的方法。 (二)实时原理 1、常规PCR技术: 对PCR扩增反应的终点产物进行定量和定性分析无法对起始模板准确定量,无法对扩增反应实时检测。 2、实时定量PCR技术: 利用荧光信号的变化实时检测PCR扩增反应中每一个循环扩增产物量的变化,通过Ct值和标准曲线的分析对起始模板进行定量分析 3、如何对起始模板定量? 通过Ct值和标准曲线对起始模板进行定量分析. 4、几个概念: (1)扩增曲线: (2)荧光阈值:
(3)Ct值: CT值的重现性:
5、定量原理: 理想的PCR反应:X=X0*2n 非理想的PCR反应:X=X0 (1+Ex)n n:扩增反应的循环次数 X:第n次循环后的产物量 X0:初始模板量 Ex:扩增效率 5、标准曲线 6、绝对定量 1)确定未知样品的C(t)值 2)通过标准曲线由未知样品的C(t)值推算出其初始量
7、DNA的荧光标记: 二、实时荧光定量PCR的几种方法介绍 方法一:SYBR Green法 (一)工作原理 1、SYBR Green 能结合到双链DNA的小沟部位 2、SYBR Green 只有和双链DNA结合后才发荧光
3、变性时,DNA双链分开,无荧光 4、复性和延伸时,形成双链DNA,SYBR Green 发荧光,在此阶段采集荧光信号。
PCR反应体系的建立及优化: 1、SYBR Green 使用浓度:太高抑制Taq酶活性,太低,荧光信号太弱,不易检测 2、Primer:引物的特异性高,否则扩增有杂带,定量不准 3、MgCl2的浓度:可以降低到1.5mM,以减少非特异性产物 4、反应Buffer 体系的优化 5、反应温度和时间参数:由酶和引物决定 6、其他与常规PCR相同 (二)应用范围 1、起始模板的测定; 2、基因型的分析; 3、融解曲线分析:可以优化PCR反应的条件,对常规PCR有指导意义,如对primer 的评价;可以区分单一引物、引物二聚体、变异产物、多种产物。 (三)优点及缺点 优点:对DNA模板没有选择性;适用于任何DNA;使用方便;不必设计复杂探针;非常灵敏;便宜。 缺点:容易与非特异性双链DNA结合,产生假阳性;但可以通过融解曲线的分析,优化反应条件;对引物特异性要求较高。
实时荧光定量PCR(RT-qPCR)完全手册 所谓的实时荧光定量PCR就是通过对PCR 扩增反应中每一个循环产物荧光信号的实时检测从而实现对起始模板定量及定性的分析。RT-qPCR是由三个步骤组成 RT-qRCR影响分析可靠性关键点(Key porint) 关键词:荧光实时实时荧光定量PCRRT-qPCRRT-PCR反转录定量PCRQRT-PCR 方法简介 所谓的实时荧光定量PCR就是通过对PCR 扩增反应中每一个循环产物荧光信号的实时检测从而实现对起始模板定量及定性的分析。在实时荧光定量PCR反应中,引入了一种荧光化学物质,随着PCR 反应的进行,PCR 反应产物不断累计,荧光信号强度也等比例增加。每经过一个循环,收集一个荧光强度信号,这样我们就可以通过荧光强度变化监测产物量的变化,从而得到一条荧光扩增曲线。 RT-qPCR是由三个步骤组成: 1.反转录:依赖反转录酶将RNA反转录成cDNDA; 2. 扩增:用PCR的方法扩增cDNA; 3.检测:实时检测和定量扩增的产物. RT-qRCR影响分析可靠性关键点(Key porint): 1.分析结果依赖于模板的数量、质量以及合理的检测方法设计 2.反转录反应的非标准化影响试验的稳定性 3.数据分析应该高度客观,如果不合理的分析,从分析结果中会得到混淆的错误结果,因此通过对RT-qPCR的每一组分进行质量评价以达到最小化变异性,最大化可重复性,而且还需要沿用一个通用的数据分析的指南。对基因表达分析的标准化的需要是与人类临床诊断分析相适应的。
存在的问题 由于各个学术团体和科研机构使用不同的操作流程,必然导致大家使用不同定量的来源物以及数据分析: 1.新鲜、冰冻、甲醛固定的样品 2.整个组织样本,显微切割样本,单个细胞,组培细胞 3.总RNA或者mRNA 4.RNA反转录成cDNA的不同的引发策略 5.不同的酶以及酶的不同组合 6.变异系数、灵敏度 7. 多类型的检测化学方法,反应的条件,热循环仪的分析以及汇报方式。 8.每一步骤缺乏标准化分析流程造成了在样品的处理,内参的使用,归一化的方法,质量控制等等因素严重影响RT-qPCR的可信度,重复性。 RNA 质量评价 现在RNA 定量的程序很多。最近EMBO qPCR course (http://www-db.embl.de/jss/Embl GroupsOrg/conf_28) 比较了用Ribogreen, Agilent BioAnalyser, spectrophotometer,Nan odrop and the BioRadExperion来定量同样的样品。结果显示没有哪两种方法得到同样的
以下实验步骤仅供参考: 1 样品RNA的抽提 ①取冻存已裂解的细胞,室温放置5分钟使其完全溶解。 ②两相分离每1ml的TRIZOL试剂裂解的样品中加入0.2ml的氯仿,盖紧管盖。手动剧烈振荡管体15秒后,15到30℃孵育2到3分钟。4℃下12000rpm离心15分钟。离心后混合液体将分为下层的红色酚氯仿相,中间层以及无色水相上层。RNA全部被分配于水相中。水相上层的体积大约是匀浆时加入的TRIZOL 试剂的60%。 ③RNA沉淀将水相上层转移到一干净无RNA酶的离心管中。加等体积异丙醇混合以沉淀其中的RNA,混匀后15到30℃孵育10分钟后,于4℃下12000rpm 离心10分钟。此时离心前不可见的RNA沉淀将在管底部和侧壁上形成胶状沉淀块。 ④RNA清洗移去上清液,每1mlTRIZOL试剂裂解的样品中加入至少1ml的75%乙醇(75%乙醇用DEPCH2O配制),清洗RNA沉淀。混匀后,4℃下7000rpm 离心5分钟。 ⑤RNA干燥小心吸去大部分乙醇溶液,使RNA沉淀在室温空气中干燥5-10分钟。 ⑥溶解RNA沉淀溶解RNA时,先加入无RNA酶的水40μl用枪反复吹打几次,使其完全溶解,获得的RNA溶液保存于-80℃待用。 2 RNA质量检测 1)紫外吸收法测定 先用稀释用的TE溶液将分光光度计调零。然后取少量RNA溶液用TE稀释(1:100)后,读取其在分光光度计260nm和280nm处的吸收值,测定RNA 溶液浓度和纯度。 ①浓度测定 A260下读值为1表示40 μg RNA/ml。样品RNA浓度(μg/ml)计算公式为:A260 ×稀释倍数× 40 μg/ml。具体计算如下: RNA溶于40 μl DEPC水中,取5ul,1:100稀释至495μl的TE中,测得A260 = 0.21 RNA 浓度= 0.21 ×100 ×40 μg/ml = 840 μg/ml 或0.84 μg/μl 取5ul用来测量以后,剩余样品RNA为35 μl,剩余RNA总量为: 35 μl × 0.84 μg/μl = 29.4 μg ②纯度检测 RNA溶液的A260/A280的比值即为RNA纯度,比值范围1.8到2.1。 2)变性琼脂糖凝胶电泳测定 ①制胶 1g琼脂糖溶于72ml水中,冷却至60℃,10 ml的10× MOPS电泳缓冲液和18 ml的37% 甲醛溶液(12.3 M)。 10×MOPS电泳缓冲液 浓度成分 0.4M MOPS,pH 7.0 0.1M 乙酸钠 0.01M EDTA 灌制凝胶板,预留加样孔至少可以加入25 μl溶液。胶凝后取下梳子,将凝胶板
实时荧光定量pcr步骤: 荧光定量PCR 实验步骤:①取冻存已裂解的细胞,室温放置5分钟使其完全溶解。②两相分离每1ml的TRIZOL试剂裂解的样品中加入0.2ml的氯仿,盖紧管盖。手动剧烈振荡管体15秒后,15到30℃孵育2到3分钟。4℃下12000rpm离心15分钟。离心后混合液体将分为下层的红色酚氯仿相,中间层以及无色水相上层。RNA全部被分配于水相中。水相上层的体积大约是匀浆时加入的TRIZOL试剂的60%。③RNA沉淀将水相上层转移到一干净无RNA 酶的离心管中。加等体积异丙醇混合以沉淀其中的RNA,混匀后15到30℃孵育10分钟后,于4℃下12000rpm 离心10分钟。此时离心前不可见的RNA沉淀将在管底部和侧壁上形成胶状沉淀块。 ④RNA清洗移去上清液,每1mlTRIZOL试剂裂解的样品中加入至少1ml的75%乙醇(75%乙醇用DEPCH2O配制),清洗RNA沉淀。混匀后,4℃下7000rpm离心5分钟。⑤RNA干燥小心吸去大部分乙醇溶液,使RNA沉淀在室温空气中干燥5-10分钟。⑥溶解RNA 沉淀溶解RNA时,先加入无RNA酶的水40μl用枪反复吹打几次,使其完全溶解,获得的RNA溶液保存于-80℃待用。1)紫外吸收法测定先用稀释用的TE溶液将分光光度计调零。然后取少量RNA 溶液用TE稀释(1:100)后,读取其在分光光度计260nm和280nm 处的吸收值,测定RNA溶液浓度和纯度。① 浓度测定A260下读值为1表示40 μg RNA/ml。样品RNA浓度(μg/ml)计算公式为:A260 ×稀释倍数× 40 μg/ml。具体计算如下:RNA溶于40 μl DEPC
利用实时定量PCR和2-△△CT法分析基因相对表达量 METHODS 25, 402–408 (2001) Analysis of Relative Gene Expression Data Using Real-Time Quantitati ve PCR and the 2-△△CT Method Kenneth J. Livak* and Thomas D. Schmittgen?,1 *Applied Biosystems, Foster City, California 94404; and ? Department of Pharmaceutical Sciences, College of Pharmacy, Washington State University, Pullman, Washington 99164-6534 摘要: 现在最常用的两种分析实时定量PCR 实验数据的方法是绝对定量和相对定量。绝对定量通过标准曲线计算起始模板的拷贝数;相对定量方法则是比较经过处理的样品和未经处理的样品目标转录本之间的表达差异。2-△△CT方法是实时定量P CR 实验中分析基因表达相对变化的一种简便方法,即相对定量的一种简便方法。本文介绍了该方法的推导,假设及其应用。另外,在本文中我们还介绍了两种2-△△CT衍生方法的推导和应用,它们在实时定量 PCR 数据分析中可能会被用到。 关键词:反转录PCR 定量PCR 相对定量实时PCR Taqman 反转录 PCR (RT-PCR )是基因表达定量非常有用的一种方法(1 - 3 )。实时PCR 技术和RT-PCR 的结合产生了反转录定量 PCR 技术(4 ,5 )。实时定量 P CR 的数据分析方法有两种:绝对定量和相对定量。绝对定量一般通过定量标准曲线来确定我们所感兴趣的转录本的拷贝数;相对定量方法则是用来确定经过不同处理的样品目标转录本之间的表达差异或是目标转录本在不同时相的表达差异。 绝对定量通常在需要确定转录本绝对拷贝数的条件下使用。通过实时 PCR 进行绝对定量已有多篇报道(6 - 9 ),包括已发表的两篇研究论文(10,11 )。在有些情况下,并不需要对转录本进行绝对定量,只需要给出相对基因表达差异即可。显然,我们说 X 基因在经过某种处理後表达量增加 2.5 倍比说该基因的表达从1000 拷贝/ 细胞增加到2500 拷贝/ 细胞更加直观。
定量PCR技术 ①传统终点定量法:为终点定量,重现性差、误差较大,只能作半定量、粗略定量 缺点:1、传统定量方法都是终点检测,即PCR到达平台期后进行检测,而PCR经过对数期扩增到达平台期时,检测重现性极差,无法直接从终点产物量推算出起始模板量。加入内标后,可部分消除终产物定量所造成的不准确性。 2、在待测样品中加入内标,则PCR反应变为双重PCR,双重PCR反应中存在两种模板之间的干扰和竞争,尤其当两种模板的 起始拷贝数相差比较大时,这种竞争会表现得更为显著,由于待测样品的起始拷贝数是未知的,所以无法加入合适数量 的已知模板作为内标。 方法:以看家基因为内参,通过目的基因与看家基因扩增产物的电泳条带的灰度、宽窄、光密度比值来确定待测目的基因的表达量。 内参基因(看家基因):在细胞中的表达量或在基因组中拷贝数恒定,受环境因素影响较小,用于对样本初始浓度差异进行均一化校正,常用的有β-actin、GAPDH、18sRNA等。 1)内参照法:在不同的PCR反应管中加入已定量的内标和引物,内标用基因工程方法合成。上游引物用荧光标记,下游引物不标记。在模板扩增的同时,内标也被扩增。在PCR产物中,由于内标与靶模板的长度不同,二者的扩增产物可用电泳或高效液相分离开来,分别测定其荧光强度,以内标为对照定量待检测模板。 2)竞争法:选择由突变克隆产生的含有一个新内切位点的外源竞争性模板。在同一反应管中,待测样品与竞争模板用同一对引物同时扩增(其中一个引物为荧光标记)。扩增后用内切酶消化PCR产物,竞争性模板的产物被酶解为两个片段,而待测模板不被酶切,可通过电泳或高效液相将两种产物分开,分别测定荧光强度,根据已知模板推测未知模板的起始拷贝数。 3)PCR-ELISA法:利用地高辛或生物素等标记引物,扩增产物被固相板上特异的探针所结合,再加入抗地高辛或生物素酶标抗体-辣根过氧化物酶结合物,最终酶使底物显色。常规的PCR-ELISA法只是定性实验,若加入内标,作出标准曲线,也可实现定量检测目的。 ②荧光实时定量法:为扩增期定量,重现性好、误差较小 SYBR GreenⅠ染料法 TaqMan Probe探针法 1、绝对定量:标准曲线 扩增曲线:荧光强度–循环数曲线→ Ct值 标准曲线:初始模板量对数– Ct值(呈线性相关) 数学基础:理想状态 X n = X0 × 2n 非理性状态:X n = X0(1 + Ex)n Ex代表扩增效率 →logX n = log(X0(1 + Ex)n) →logX0 = (-log(1 + Ex))n + logX n →logX0 = (-log(1 + Ex))n + logX Ct X ct为达Ct值时扩增量 方法:将已知浓度的相应DNA模板(标准品)梯度稀释,根据实时PCR得到相应的Ct值,构建标准曲线;同时,待测样品也进行PCR,得到未知浓度的待测样品的Ct值,再根据标准曲线得出其初始浓度。 标准品:a.含有和待测样品相同的扩增片段的克隆质粒 b.含有和待测样品相同的扩增片段的cDNA c.纯化的待测样品PCR扩增产物 紫外分光光度计或荧光酶标测定标准样品浓度,根据质粒或cDNA分子量换算成拷贝数,作系列梯度稀释。 待测样品浓度 = OD260× 40 ×稀释倍数(ng/μl) 待测样品分子量 = 碱基数× 324 待测样品拷贝数 = 待测样品浓度/待测样品分子量× 6 × 1014
实时荧光定量PCR操作步骤 以下实验步骤仅供参考: 1 样品RNA的抽提 ①取冻存已裂解的细胞,室温放臵5分钟使其完全溶解。 ②两相分离每1ml的TRIZOL试剂裂解的样品中加入0.2ml的氯仿,盖紧管盖。手动剧烈振荡管体15秒后,15到30℃孵育2到3分钟。4℃下12000rpm离心15分钟。离心后混合液体将分为下层的红色酚氯仿相,中间层以及无色水相上层。RNA全部被分配于水相中。水相上层的体积大约是匀浆时加入的TRIZOL试剂的60%。 ③RNA沉淀将水相上层转移到一干净无RNA酶的离心管中。加等体积异丙醇混合以沉淀其中的RNA,混匀后15到30℃孵育10分钟后,于4℃下12000rpm 离心10分钟。此时离心前不可见的RNA沉淀将在管底部和侧壁上形成胶状沉淀块。 ④RNA清洗移去上清液,每1mlTRIZOL试剂裂解的样品中加入至少1ml的75% O配制),清洗RNA沉淀。混匀后,4℃下7000rpm离心乙醇(75%乙醇用DEPCH 2 5分钟。 ⑤RNA干燥小心吸去大部分乙醇溶液,使RNA沉淀在室温空气中干燥5-10分钟。 ⑥溶解RNA沉淀溶解RNA时,先加入无RNA酶的水40μl用枪反复吹打几次,使其完全溶解,获得的RNA溶液保存于-80℃待用。 2 RNA质量检测 1)紫外吸收法测定 先用稀释用的TE溶液将分光光度计调零。然后取少量RNA溶液用TE稀释(1:100)后,读取其在分光光度计260nm和280nm处的吸收值,测定RNA溶液浓度和纯度。 ①浓度测定 A260下读值为1表示40 μg RNA/ml。样品RNA浓度(μg/ml)计算公式为:A260 ×稀释倍数× 40 μg/ml。具体计算如下: RNA溶于40 μl DEPC水中,取5ul,1:100稀释至495μl的TE中,测得A260 = 0.21 RNA 浓度= 0.21 ×100 ×40 μg/ml = 840 μg/ml 或 0.84 μg/μl 取5ul用来测量以后,剩余样品RNA为35 μl,剩余RNA总量为: 35 μl × 0.84 μg/μl = 29.4 μg ②纯度检测 RNA溶液的A260/A280的比值即为RNA纯度,比值范围1.8到2.1。 2)变性琼脂糖凝胶电泳测定 ①制胶 1g琼脂糖溶于72ml水中,冷却至60℃,10 ml的10× MOPS电泳缓冲液和18 ml 的37% 甲醛溶液(12.3 M)。 10×MOPS电泳缓冲液 浓度成分 0.4M MOPS,pH 7.0 0.1M乙酸钠 0.01M EDTA 灌制凝胶板,预留加样孔至少可以加入25 μl溶液。胶凝后取下梳子,将凝胶板
实时荧光定量PCR (Quantitative Real-time PCR)是一种在DNA扩增反应中,以荧光化学物质测每次聚合酶链式反应(PCR)循环后产物总量的方法。通过内参或者外参法对待测样品中的特定DNA序列进行定量分析的方法。· Real-timePCR是在PCR扩增过程中,通过荧光信号,对PCR 进程进行实时检测。由于在PCR扩增的指数时期,模板的Ct值和该模板的起始拷贝数存在线性关系,所以成为定量的依据。 ①取冻存已裂解的细胞,室温放置5分钟使其完全溶解。 ②两相分离每1ml的TRIZOL试剂裂解的样品中加入0.2ml的氯仿,盖紧管盖。手动剧烈振荡管体15秒后,15到30℃孵育2到3分钟。4℃下12000rpm离心15分钟。离心后混合液体将分为下层的红色酚氯仿相,中间层以及无色水相上层。RNA全部被分配于水相中。水相上层的体积大约是匀浆时加入的TRIZOL试剂的60%。 ③RNA沉淀将水相上层转移到一干净无RNA酶的离心管中。加等体积异丙醇混合以沉淀其中的RNA,混匀后15到30℃孵育10分钟后,于4℃下12000rpm 离心10分钟。此时离心前不可见的RNA沉淀将在管底部和侧壁上形成胶状沉淀块。 ④RNA清洗移去上清液,每1mlTRIZOL试剂裂解的样品中加入至少1ml的75%乙醇(75%乙醇用DEPCH2O配制),清洗RNA 沉淀。混匀后,4℃下7000rpm离心5分钟。 ⑤RNA干燥小心吸去大部分乙醇溶液,使RNA沉淀在室温空气中干燥5-10分钟。
⑥溶解RNA沉淀溶解RNA时,先加入无RNA酶的水40μl 用枪反复吹打几次,使其完全溶解,获得的RNA溶液保存于-80℃待用。
一、HBV DNA荧光定量PCR技术 (一)标本 血清、血浆(其它如组织细胞、乳汁、精子、尿液等体液);标本应封闭保存,特别在夏季,如果标本暴露的过久,开盖保存,很容易出现水分蒸发,检测结果和上一次的检测结果会有较大的差异。环境条件影响不大,比如保存在 -8°、 -20°、 24°、还是室温,对高中低病毒含量的最后的检测结果影响不是太大,只要封闭保存就可以。因此可常温运输,但不可泄露。 (二)检测方法 国内最常使用 PEG (聚乙二醇方法)法进行核酸提取,采用 TaqMan 探针进行荧光PCR 扩增。扩增 40 循环进行结果分析,如下图一所示:典型的双 S 形的曲线图,越早出现荧光曲线的病毒含量越高,越晚越低。根据不同的梯度的四个标准品进行定量。如果血清或标本里面没有病毒,就是一个阴性直线。最近我们在彻底解决污染问题之后采用扩增 50 个循环的方法。优点是让扩增管里的反应液充分反应,缺点是如果实验室存在一个潜在的污染或操作人员操作习惯不好,很容易出现携带污染,造成假阳性。对实验室的来说不应该通过减少扩增的循环数来解决污染问题,而应该通过科学的管理,提高试剂的质量,以及养成一个良好的操作习惯,或防污染的习惯来解决假阳性的问题。增加循环数有利于标本检测结果的判断,这将来可能是实验室发展的一个趋向。 COBAS TaqMan 技术在我们国家已经有好多实验室有这种全自动的核酸提取扩增系统,它的优势重点在于检测病毒含量比较低的血清标本,但是对于病毒含量比较高的,往往出现荧光 PCR 竞争的缺点。 图一为: PCR 扩增曲线
(三)污染的预防与排除 PCR 扩增产物污染是 PCR 反应中最主要最常见的污染问题,所以扩增区的仪器如枪头等要注意。最可能造成 PCR 产物污染的形式是气溶胶污染;在空气与液体面摩擦时就可形成气溶胶,在操作时比较剧烈地摇动反应管,开盖时、吸样时及污染进样枪的反复吸样都可形成气溶胶而污染。据计算一个气溶胶颗粒可含 48000 拷贝 , 因而由其造成的污染是一个值得特别重视的问题。标本处理区,包括扩增摸板的制备; PCR 扩增区,包括反应液的配制和 PCR 扩增;产物分析区,凝胶电泳分析,产物拍照及重组克隆的制备。各工作区要有一定的隔离,操作器材专用,要有一定的方向性。如:标本制备→PCR 扩增→产物分析→产物处理。切记 : 产物分析区的产物及器材不要拿到其他两个工作区。使用一次性吸头,严禁与 PCR 产物分析室的吸头混用,吸头不要长时间暴露于空气中,避免气溶胶的污染。 (四)常用仪器 常用的 PCR 仪器: ABI 系列、罗氏系列、安捷伦系列、国产 SLAN 等。 (五)临床意义 检测血清或血浆 HBV DNA 阳性有什么临床意义? 1. 判断血清中是否有 HBV DNA 的存在 我们检测到的 HBV DNA 包括完整 HBV 病毒和 DNA 片段。目前我们用的 HBV DNA 荧光 PCR 法,还不能区分是完整病毒还是片段。只要检测到的病毒载量在检测线以上就可以定为标本含有 HBV DNA 。 2. 判断抗病毒疗效的评估指标
半定量和实时定量PCR步骤 实验原理: RT-PCR将以RNA为模板的cDNA合成同PCR结合在一起,提供了一种分析基因表达的快速灵敏的方法。RT-PCR用于对表达信息进行检测或定量。另外,这项技术还可以用来检测基因表达差异或不必构建cDNA文库克隆cDNA。RT-PCR比其他包括Northern印迹、RNase 保护分析、原位杂交及S1核酸酶分析在内的RNA分析技术,更灵敏,更易于操作。 RT-PCR的模板可以为总RNA或poly(A)+选择性RNA。逆转录反应可以使用逆转录酶,以随机引物、oligo(dT)或基因特异性的引物(GSP)起始。RT-PCR可以一步法或两步法的形式进行。在两步法RT-PCR中,每一步都在最佳条件下进行。cDNA的合成首先在逆转录缓冲液中进行,然后取出1/10的反应产物进行PCR。在一步法RT-PCR中,逆转录和PCR在同时为逆转录和PCR优化的条件下,在一只管中顺次进行。 实验步骤: Trizol法RNA提取步骤 1、提取总RNA 2、逆转录反应 3、PCR反应以下实验步骤仅供参考: 1 样品RNA的抽提 ①取冻存已裂解的细胞,室温放置5分钟使其完全溶解。 ②两相分离每1ml的TRIZOL试剂裂解的样品中加入0.2ml的氯仿,盖紧管盖。手动剧烈振荡管体15秒后,15到30℃孵育2到3分钟。4℃下12000rpm离心15分钟。离心后混合液体将分为下层的红色酚氯仿相,中间层以及无色水相上层。RNA全部被分配于水相中。水相上层的体积大约是匀浆时加入的TRIZOL试剂的60%。 ③RNA沉淀将水相上层转移到一干净无RNA酶的离心管中。加等体积异丙醇混合以沉淀其中的RNA,混匀后15到30℃孵育10分钟后,于4℃下12000rpm 离心10分钟。此时离心前不可见的RNA沉淀将在管底部和侧壁上形成胶状沉淀块。 ④RNA清洗移去上清液,每1mlTRIZOL试剂裂解的样品中加入至少1ml的75%乙醇(75%乙醇用DEPCH2O配制),清洗RNA沉淀。混匀后,4℃下7000rpm离心5分钟。⑤RNA 干燥小心吸去大部分乙醇溶液,使RNA沉淀在室温空气中干燥5-10分钟。 ⑥溶解RNA沉淀溶解RNA时,先加入无RNA酶的水40μl用枪反复吹打几次,使其完全溶解,获得的RNA溶液保存于-80℃待用。 2 RNA质量检测 1)紫外吸收法测定 先用稀释用的TE溶液将分光光度计调零。然后取少量RNA溶液用TE稀释(1:100)后,读取其在分光光度计260nm和280nm处的吸收值,测定RNA溶液浓度和纯度。①浓度测定 A260下读值为1表示40 μg RNA/ml。样品RNA浓度(μg/ml)计算公式为:A260 ×稀释倍数× 40 μg/ml。具体计算如下: RNA溶于40 μl DEPC水中,取5ul,1:100稀释至495μl的TE中,测得A260 = 0.21 RNA 浓度= 0.21 ×100 ×40 μg/ml = 840 μg/ml 或 0.84 μg/μl 取5ul用来测量以后,剩余样品RNA为35 μl,剩余RNA总量为: 35 μl × 0.84 μg/μl = 29.4 μg ②纯度检测 RNA溶液的A260/A280的比值即为RNA纯度,比值范围1.8到2.1。 2)变性琼脂糖凝胶电泳测定①制胶 1g琼脂糖溶于72ml水中,冷却至60℃,10 ml的10× MOPS电泳缓冲液和18 ml的37% 甲醛溶液(12.3 M)。 10×MOPS电泳缓冲液浓度成分 0.4M MOPS,pH 7.0 0.1M 乙酸钠 0.01M EDTA 灌制凝胶板,预留加样孔至少可以加入25 μl溶液。胶凝后取下梳子,将凝胶板放入电泳槽内,加足量的1×MOPS电泳缓冲液至覆盖胶面几个毫米。②准备RNA样品