
第九章光谱分析法概论- 章节小结
1.基本概念
电磁辐射:是一种以巨大速度通过空间而不需要任何物质作为传播媒介的光子流。
磁辐射性质:波动性、粒子性
电磁波谱:所有的电磁辐射在本质上是完全相同的,它们之间的区别仅在于波长或频率不同。若把电磁辐射按波长长短顺序排列起来,即为电磁波谱。
光谱和光谱法:当物质与辐射能相互作用时,物质内部发生能级跃迁,记录由能级跃迁所产生的辐射能强度随波长(或相应单位)的变化,所得的图谱称为光谱。利用物质的光谱进行定性、定量和结构分析的方法称光谱法。
非光谱法:是指那些不以光的波长为特征讯号,仅通过测量电磁辐射的某些基本性质(反射、折射、干涉、衍射和偏振)的变化的分析方法。
原子光谱法:测量气态原子或离子外层电子能级跃迁所产生的原子光谱为基础的成分分析方法。为线状光谱。
分子光谱法:以测量分子转动能级、分子中原子的振动能级(包括分子转动能级)和分子电子能级(包括振-转能级跃迁)所产生的分子光谱为基础的定性、定量和物质结构分析方法。为带状光谱。
吸收光谱法:物质吸收相应的辐射能而产生的光谱,其产生的必要条件是所提供的辐射能量恰好满足该吸收物质两能级间跃迁所需的能量。利用物质的吸收光谱进行定性、定量及结构分析的方法称为吸收光谱法。
发射光谱法:发射光谱是指构成物质的原子、离子或分子受到辐射能、热能、电能或化学能的激发跃迁到激发态后,由激发态回到基态时以辐射的方式释放能量,而产生的光谱。利用物质的发射光谱进行定性定量及结构分析的方法称为发射光谱法。
2.基本计算
(1)电磁辐射的频率:ν=C/λ σ=1/λ=ν/C
(2)电磁辐射的能量:E=hν=hC/λ=hCσ
3.光谱分析仪器组成:辐射源、分光系统、检测系统
第十章紫外-可见分光光度法- 章节小结
1.基本概念
透光率(T):透过样品的光与入射光强度之比。T=I t/I0
吸光度(A):透光率的负对数。A=-lgT=lg(I0/I t)
吸光系数(E):吸光物质在单位浓度及单位厚度时的吸光度。根据浓度单位的不同,常有摩尔吸光系数ε和百分吸光系数之分。
电子跃迁类型:
(1)ζ-ζ*跃迁:处于ζ成键轨道上的电子吸收光能后跃迁到ζ*反键轨道。饱和烃中电子跃迁均为此种类型,吸收波长小于150nm。
(2)π-π*跃迁:处于π成键轨道上的电子吸收光能后跃迁到π*反键轨道上,所需的能量小于ζ-ζ*跃迁所需的能量。孤立的π-π*跃迁吸收波长一般在200nm左右,共轭的π-π*跃迁吸收波长
>200nm,强度大。
(3)n-π*跃迁:含有杂原子不饱和基团,其非键轨道中的孤对电子吸收能量后向π*反键轨道跃迁,这种吸收一般在近紫外区(200-400nm),强度小。
(4)n-ζ*跃迁:含孤对电子的取代基,其杂原子中孤对电子吸收能量后向ζ*反键轨道跃迁,吸收波长约在200nm。
以上四种类型跃迁所需能量ζ-ζ* > n-ζ*≥ π-π* > n-π*
(5)电荷迁移跃迁和配位场跃迁
生色团:有机化合物分子结构中含有π-π*或n-π*跃迁的基团,能在紫外-可见光范围内产生吸收的原
子团。
助色团:含有非键电子的杂原子饱和基团,与生色团或饱和烃连接时,能使该生色团或饱和烃的吸收峰向长波方向移动,并使吸收强度增加的基团。
红移(长移):由于化合物的结构改变,如发生共轭作用、引入助色团以及溶剂改变等,使吸收峰向长波方向移动。
蓝移(紫移或短移):当化合物的结构改变或受溶剂影响使吸收峰向短波方向移动。
增色效应:由于化合物结构改变或其他原因,使吸收强度增加。
减色效应:由于化合物结构改变或其他原因,使吸收强度减小。
强带:化合物的紫外可见吸收光谱中,摩尔吸光系数值大于104的吸收峰。
弱带:化合物的紫外可见吸收光谱中,摩尔吸光系数值小于102的吸收峰。
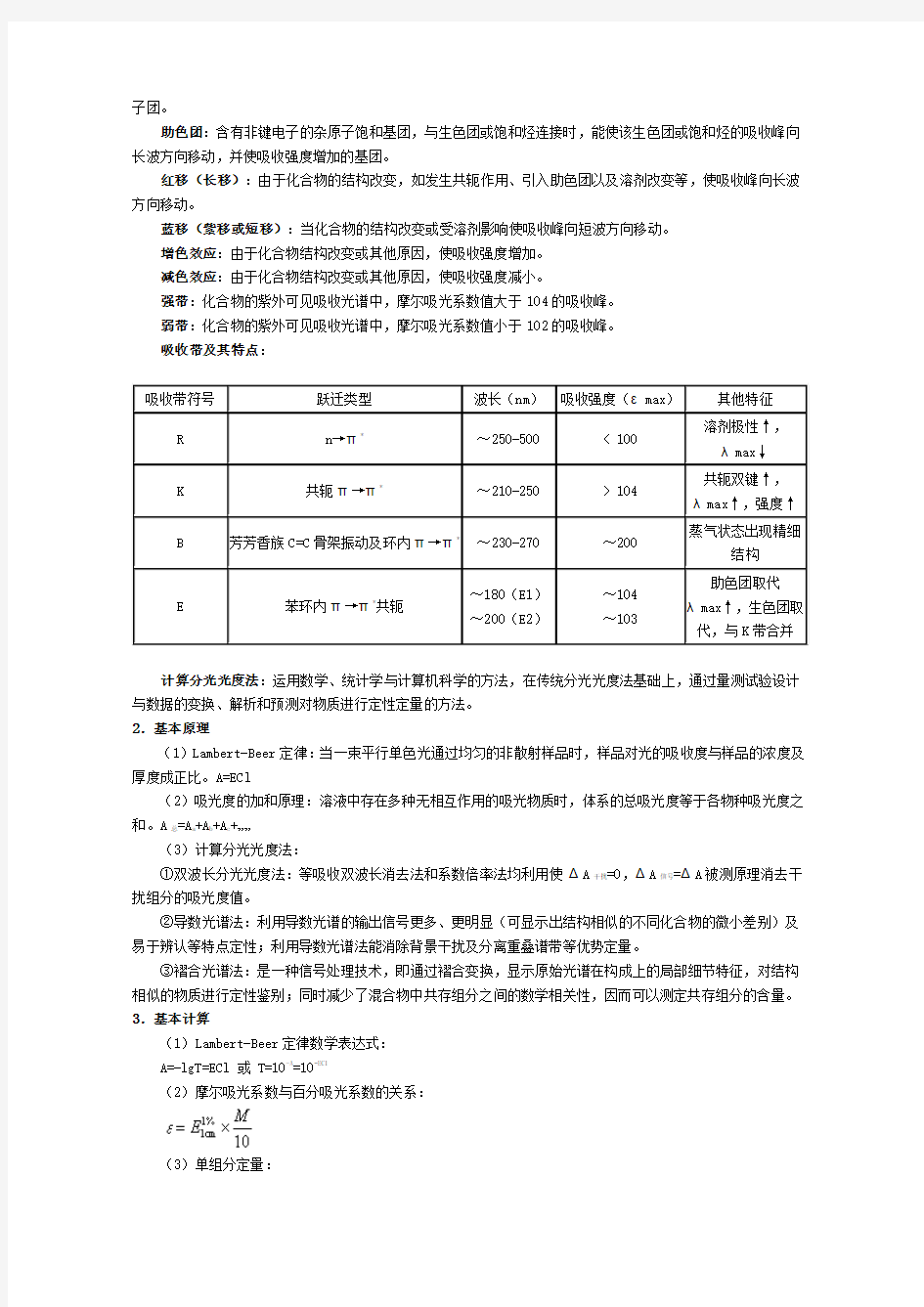
吸收带及其特点:
计算分光光度法:运用数学、统计学与计算机科学的方法,在传统分光光度法基础上,通过量测试验设计与数据的变换、解析和预测对物质进行定性定量的方法。
2.基本原理
(1)Lambert-Beer定律:当一束平行单色光通过均匀的非散射样品时,样品对光的吸收度与样品的浓度及厚度成正比。A=ECl
(2)吸光度的加和原理:溶液中存在多种无相互作用的吸光物质时,体系的总吸光度等于各物种吸光度之和。A总=A a+A b+A c+……
(3)计算分光光度法:
①双波长分光光度法:等吸收双波长消去法和系数倍率法均利用使ΔA干扰=0,ΔA信号=ΔA被测原理消去干扰组分的吸光度值。
②导数光谱法:利用导数光谱的输出信号更多、更明显(可显示出结构相似的不同化合物的微小差别)及易于辨认等特点定性;利用导数光谱法能消除背景干扰及分离重叠谱带等优势定量。
③褶合光谱法:是一种信号处理技术,即通过褶合变换,显示原始光谱在构成上的局部细节特征,对结构相似的物质进行定性鉴别;同时减少了混合物中共存组分之间的数学相关性,因而可以测定共存组分的含量。3.基本计算
(1)Lambert-Beer定律数学表达式:
A=-lgT=ECl 或 T=10-A=10-ECl
(2)摩尔吸光系数与百分吸光系数的关系:
(3)单组分定量:
① 吸光系数法:C=A/El
② 对照法:
③ 校正曲线法
(4)多组分定量(a + b的混合物):
①解线性方程组:
② 等吸收双波长消去法:
③系数倍率法:ΔA=
第十一章荧光分析法- 章节小结
1.基本概念:
荧光:物质分子接受光子能量而被激发,然后从激发态的最低振动能级返回基态时发射出的光称为荧光。
振动弛豫:物质分子吸收能量后,跃迁到电子激发态的几个振动能级上。激发态分子通过与溶剂分子的碰撞而将部分振动能量传递给溶剂分子,其电子则返回到同一电子激发态的最低振动能级的过程。
内部能量转换(简称内转换):当两个电子激发态之间的能量相差较小以致其振动能级有重叠时,受激分子常由高电子能级以无辐射方式转移至低电子能级的过程。
荧光发射:处于激发单重态的分子,通过内转换及振动弛豫,返回到第一激发单重态的最低振动能级,然后再以辐射形式发射光量子而返回至基态的任一振动能级上,这时发射的光量子称为荧光。
外部能量转换(简称外转换):在溶液中激发态分子与溶剂分子及其他溶质分子之间相互碰撞而以热能的形式放出能量的过程。
体系间跨越:在某些情况下处于激发态分子的电子发生自旋反转而使分子的多重性发生变化,分子由激发单重态跨越到激发三重态的过程。
磷光发射:经过体系间跨越的分子再通过振动弛豫降至激发三重态的最低振动能级,分子在此三重态的最低振动能级存活一段时间后返回至基态的各个振动能级而发出的光辐射。
激发光谱:是荧光强度(F)对激发波长(λex)的关系曲线,它表示不同激发波长的辐射引起物质发射某一波长荧光的相对效率。
发射光谱(称荧光光谱):是荧光强度(F)对发射波长(λem)的关系曲线,它表示当激发光的波长和强度保持不变时,在所发射的荧光中各种波长组分的相对强度。
2.基本原理
(1)荧光是物质分子接受光子能量被激发后,从激发态的最低振动能级返回基态时发射出的光。荧光分析法具有灵敏度高、选择性好的优点。
(2)荧光光谱具有如下特征:荧光波长总是大于激发光波长、荧光光谱的形状与激发波长无关、荧光光谱与激发光谱存在“镜像对称”关系。
(3)能够发射荧光的物质应同时具备的两个条件:物质分子必须有强的紫外-可见吸收;物质分子必须有一定的荧光效率。
(4)在荧光法测定时常有散射光存在,主要有瑞利散射和拉曼散射。瑞利散射:光子和物质分子发生弹性碰撞,不发生能量的交换,仅仅是光子运动方向发生改变,其波长与入射光波长相同。拉曼散射:光
子和物质分子发生非弹性碰撞时,在光子运动方向发生改变的同时,光子与物质分子发生能量的交换,发射出比入射光稍长或稍短的光。波长比入射光更长的拉曼散射光对荧光测定有干扰。采取一定的措施(如选择适当的波长或溶剂)可消除拉曼光的干扰。
(5)荧光法常被用于定性和定量分析,但其定量应用更为广泛。荧光分析法定量的依据是:当ECL≤0.05时,F=2.3K’I0ECl=KC。可采用的定量分析方法有:标准曲线法、比例法、联立方程式法。
(6)用于荧光法测定的仪器是荧光分光光度计,其主要部件包括:激发光源、激发单色器(置于样品池前)和发射单色器(置于样品池后)、样品池及检测系统。
(7)为进一步提高荧光分析法灵敏度和选择性,发展了其他的荧光分析技术,主要有激光荧光分析、时间分辨荧光和同步荧光分析等。
第十二章红外吸收光谱法 - 章节小结1.基本概念
基频峰:当分子吸收一定频率的红外线,由振动基态(V=0)跃迁至第一激发态(V=1)时,所产生的吸收峰称为基频峰。
泛频峰:将倍频峰、合频峰及差频峰统称为泛频峰。
伸缩振动:化学键两端的原子沿着键轴方向作规律性的伸缩运动。
弯曲振动:键角发生规律性变化的振动,又称为变形振动。
振动自由度:分子基本振动的数目。
简并:振动形式不同但振动频率相同而合并的现象称为简并。
红外活性振动:能引起偶极矩变化而吸收红外线的振动。
红外非活性振动:不能引起偶极矩变化,不吸收红外线的振动。
特征峰:凡是能用于鉴别基团存在的吸收峰。
相关峰:由一个基团产生的一组相互具有依存关系的吸收峰。
特征区:4000~1300cm-1的区域称为特征区。
指纹区:1300~400cm-1区域称为指纹区。
2.基本原理
(1)振动自由度:非线型分子有3N-6个振动自由度;线型分子有3N-5个。
(2)红外吸收光谱产生的条件:①E
L =ΔV·hγ或γ
L
=ΔV·γ;②Δμ≠0。
(3)基频峰的分布规律:①μ愈小,ζ愈高。②μ相同,K愈大,ζ愈高。③μ相同时,一般ν>β>γ。
(4)解析光谱的三大要素:第一是峰位,第二是峰强,第三是峰形。
(5)解析光谱的原则:遵循用一组相关峰确定一个基团。
(6)解析光谱的顺序:先特征区,再指纹区。
(7)掌握各类化合物的主要光谱特征。
3.基本计算
①②γ
L
=ΔV·γ
③④
第十三章原子吸收分光光度法 - 章节小结1.基本概念
共振吸收线:原子从基态激发到能量最低的激发态(第一激发态)产生的谱线。
半宽度:原子吸收线中心频率(ν0)的吸收系数一半处谱线轮廓上两点之间的频率差。
积分吸收:吸收线轮廓所包围的面积,即气态原子吸收共振线的总能量。
峰值吸收:通过测量中心频率处的吸收系数来测定吸收度和原子总数。
光谱项、原子能级图、空心阴极灯、原子化器、特征浓度、特征质量。
2.基本原理
(1)原子吸收光谱分析法是基于原子蒸气对同种元素特征谱线的共振吸收作用来进行定量分析的方法。
(2)吸收线轮廓是指具有一定频率范围和形状的谱线,它可用谱线的半宽度来表征。吸收线轮廓是由自然变宽、热变宽、压力变宽等原子本身的性质和外界因素影响而产生的。
(3)采用测量峰值吸收的方法来代替测量积分吸收,必须满足以下条件:①发射线轮廓小于吸收线轮廓;②发射线与吸收线频率的中心频率重合。
(4)原子吸收光谱分析法的定量关系式:A=KC,常用的方法有:校正曲线法、标准加入法、内标法等。
(5)在原子吸收分光光度法中,干扰效应主要有:电离干扰、物理干扰、光学干扰及非吸收线干扰(背景干扰)、化学干扰等。消除方法有:加入缓冲剂、保护剂、消电离剂、配位剂等;采用标准加入法和改变仪器条件(如分辨率、狭缝宽度)或背景扣除等。
3.原子吸收分光光度计主要组成:锐线光源、原子化器、分光系统和检测系统。
第十四章核磁共振波谱法 - 章节小结1.基本概念
屏蔽效应:核外电子及其他因素对抗外加磁场的现象。
局部屏蔽效应:核外成键电子云在外加磁场的诱导下,产生与外加磁场方向相反的感应磁场,使氢核实受磁场强度稍有降低的现象。
磁各向异性效应:在外加磁场作用下,由化学键产生的感应磁场使在分子中所处的空间位置不同的核屏蔽作用不同的现象。
驰豫历程:激发核通过非辐射途径损失能量而恢复至基态的过程。
化学位移:质子由于在分子中所处的化学环境不同,而有不同的共振频率。
自旋偶合:核自旋产生的核磁矩间的相互干扰。
自旋分裂:由自旋偶合引起核磁共振峰分裂的现象称为自旋-自旋分裂。
偶合常数:由自旋分裂产生的峰裂距,反映偶合作用的强弱。
磁等价:分子中一组化学等价核(化学位移相同的核)与分子中的其它任何一个核都有相同强弱的偶合,则这组核为磁等价。
13C-1H COSY谱:两坐标轴分别为13C和1H的化学位移的二维谱。
2.基本理论
(1)共振吸收条件:①γ0=γ ②Δm=±1
(2)影响化学位移的因素:①氢核邻近原子或基团的电负性越大,δ值增大。②磁各向异性效应使处于负屏蔽区的氢核δ值大,处于正屏蔽区的氢核δ值小。③氢键中质子δ增大。分子间氢键使化学位移的改变与溶剂的性质和浓度有关。
(3)自旋分裂:一级图谱的裂分一般具有如下规律:①裂分峰数目由相邻偶合氢核数目n决定,符合n+1律,多重峰峰高比为二项式展开式的系数比。I≠1/2时,符合2nI+1律。有多组偶合程度相等的1H核时,则呈现(n+ n'+…)+1个子峰;如果偶合程度不同时,则呈现(n+1)(n'+1)个子峰。②多重峰的位置,是以化学位移值为中心左右对称,并且各裂分峰间距相等。
3.基本计算
(1)进动频率(γ)与外磁场强度(H0)关系式-Larmor方程式:
(2)屏蔽效应存在时的Larmor方程式:
第十五章质谱法 - 章节小结1.基本概念及术语
质谱分析法:质谱分析法是利用多种离子化技术,将物质分子转化为离子,选择其中带正电荷的离子使其在电场或磁场的作用下,按其质荷比m/z的差异进行分离测定,从而进行物质成分和结构分析的方法。
相对丰度:以质谱中基峰(最强峰)的高度为100%,其余峰按与基峰的比例加以表示的峰强度为相对丰度,又称相对强度。
离子源:质谱仪中使被分析物质电离成离子的部分。常见的有电子轰击源EI、化学电离源CI、快原子轰击源FAB等。
分子离子:分子通过某种电离方式,失去一个外层价电子而形成带正电荷的离子,用m·+表示。
碎片离子:当分子在离子源中获得的能量超过其离子化所需的能量时,分子中的某些化学键断裂而产生的离子。
亚稳离子:离子(m1+)脱离离子源后,在飞行过程中发生裂解而形成的低质量离子(m2+),通常用m+表示。
同位素离子:质谱图中含有同位素的离子。
单纯开裂:仅一个键发生开裂并脱去一个游离基,称单纯开裂。
重排开裂:通过断裂两个或两个以上化学键,进行重新排列的开裂方式。重排开裂一般脱去一中性分子,同时发生重排,生成重排离子。
2.重点和难点
(1)离子化机理及其特点
①电子轰击电离(EI):气化后的样品分子进入离子化室后,受到由钨或铼灯丝发射并加速的电子流的轰击产生正离子。轰击电子的能量大于样品分子的电离能,使样品分子电离或碎裂。电子轰击质谱能提供有机化合物最丰富的结构信息,有较好的重现性,其裂解规律的研究也最为完善,已经建立了数万种有机化合物的标准谱图库可供检索。其主要缺点在于不适用于分析难挥发和热稳定性差的样品。
②化学电离(CI):引入一定压力的反应气进入离子化室,反应气在具有一定能量的电子流的作用下电离或者裂解。生成的离子和反应气分子进一步反应或与样品分子发生离子-分子反应,通过质子交换使样品分子电离。化学电离属于软电离方式,通常准分子离子峰强度大,易获得有关化合物基团的信息。其主要缺点是重现性较差,且不适合于难挥发、热不稳定样品的分析。
③快原子轰击(FAB):将样品分散于基质(常用甘油等高沸点溶剂)制成溶液,涂布于金属靶上送入FAB离子源中。将经强电场加速后的惰性气体中性原子束(如氙)对准靶上样品轰击。基质中存在的缔合离子及经快原子轰击产生的样品离子一起被溅射进入气相,并在电场作用下进入质量分析器。此法优点在于离子化能力强,可用于强极性、挥发性低、热稳定性差和相对分子质量大的样品及EI和CI难于得到有意义的质谱的样品。FAB比EI容易得到比较强的分子离子或准分子离子;不同于CI的一个优势在于其所得质谱有较多的碎片离子峰信息,有助于结构解析。缺点是对非极性样品灵敏度下降,而且基质在低质量数区(400以下)产生较多干扰峰。FAB是一种表面分析技术,应注意优化表面状况的样品处理过程。
值得一提的是,在FAB离子化过程中,可同时生成正负离子,这两种离子都可以用质谱进行分析。样品分子如带有强电子捕获结构,特别是带有卤原子,可以产生大量的负离子。负离子质谱已成功用于农药残留物的分析。
(2)质谱中的主要离子及其在质谱解析中的作用
①分子离子:大多数有机化合物分子通过某种电离方式,在离子源中失去一个电子而形成带正电荷的离子(z=1),即分子离子。由于确认了分子离子即可确定化合物的相对分子质量,因而分子离子峰的正确识别十分重要。由CI、FAB等软电离方式获得的准分子离子,其作用与分子离子相当。分子离子峰一般位于质谱图中质荷比的最高端,但有时最高质荷比峰不一定是分子离子峰。其原因为: M+n(n=1、2…)同位素峰可能出现在质荷比最高处;杂质峰可能出现在最高质荷比处;当样品分子的稳定性差时,分子离子峰很弱甚至不出现,此时最高质荷比的离子是碎片离峰子。
确认分子离子峰时应依据分子离子的稳定性规律及质量数的奇偶规律,即由C、H、O组成的化合物,分子离子峰的质量数是偶数;由C、H、O、N组成的化合物,含奇数个N,分子离子峰的质量数是奇数;含偶数个N,分子离子峰的质量数是偶数。凡不符合这一规律者,不是分子离子。同时还应注意最高质荷比离子与相邻离子间的质量差是否合理,必要时可改变试验条件,如降低EI电子流能量或采用CI、FAB等软
电离技术,以便观察到分子离子峰(或准分子离子峰)。
②碎片离子:分子在离子源中获得的能量超过分子离子化所需的能量时,分子离子中某些化学键发生断裂形成碎片离子。由于键断裂的位置不同,同一分子离子可产生不同质量的碎片离子,其相对丰度与键断裂的难易及化合物的结构密切相关。因此,碎片离子的峰位(m/z)及相对丰度可提供化合物的结构信息。一般说来,高丰度的碎片离子峰代表着分子中易于裂解的部分,如果有几个主要碎片,并且代表着分子中的不同部分,则可由这些碎片将化合物的骨架粗略拼凑起来,以便进行结构确证。
③亚稳离子:离子(m1+)脱离离子源后并在到达质量分析器前,由于其内能较高或相互碰撞等因素,在飞行过程中可能发生裂解而形成低质量的离子(M2),这种离子的能量比在离子源中产生的m2+离子的能量小,且不稳定,在质谱中称其为亚稳离子,通常用m+表示。亚稳离子的特点是:峰位低于m2+峰,其峰位为m+=(m2+)2/m1+。其原因是:虽然m2+离子和m+离子均系m1+离子派生的相同质量离子,但因m+离子的能量在飞行途中被中性碎片带走了一部分,故m+离子较m2+离子的能量小,其在磁场中的偏转半径R 就小于m2+离子,因此,其峰位出现在较m2+离子低的质量区;
峰弱,峰强仅为m1+峰的1%~3%;峰钝,一般可跨2~5个质量单位;质荷比一般不是整数。亚稳离子m+与母离子m1+及子离子m2+的关系为:m+=(m2+)2/m1+,由此可确定离子间的亲缘关系,有助于了解裂解规律,解析复杂质谱。需要说明的是,并非所有的裂解过程都有亚稳离子产生,因此,若没有观察到相应的亚稳离子峰,也不能说明该裂解历程不会发生。
④同位素离子:即含有同位素的离子。重质同位素与丰度最大的轻质同位素峰的峰强比,用
…表示。由于34S、37Cl及81Br的丰度比很大,因此含有S、Cl、Br的分子离子或碎片离子其M+2峰强度较大,故可根据M和M+2两个峰强比推断分子中是否含有S、Cl、Br及其原子数目。同位素峰强比可用二项式(a+b)n展开式求出。a与b为轻质及重质同位素的丰度比,n为原子数目。若采用低分辨质谱,则可通过同位素相对丰度法推导其分子式。由于同位素离子峰的相对强度与其中各元素的天然丰度及存在个数成正比,因此,利用精确测定的(m+1)+、(m+2)+、相对于m+的强度比值,可从Beynon 表(表中收载了含C、H、N、O不同组合的质量及同位素丰度比的各类有机化合物)中查出最可能的化学式.再结合其他规则,确定化合物的分子式。
(3)重排开裂机理及主要类型
质谱中的某些离子是通过断裂两个或两个以上化学键重新排列形成的,这种裂解称为重排开裂。重排开裂生成的离子称为重排离子。与单纯开裂不同,由于重排开裂时脱去一个中性分子,因此重排开裂前后离子所带电子数的奇、偶性保持不变;其质量数的奇、偶性一般也保持不变,除非重排开裂时失去了奇数个氮原子。由于离子的电子数与质量间存在一定关系,故可根据离子质量推测该离子是否由重排开裂产生,从而有助于机化合物的结构推断。
产生重排开裂的主要原因是:重排离子具有更高的稳定性;能够脱去稳定的中性分子;需要裂解的临界能较低;开裂中心在易于移动的氢附近等。
重排开裂的方式很多,其中较常见的有McLafferty重排(麦氏重排)和逆Diels-Alder重排(RDA重排)。
①McLafferty重排:当化合物中含有不饱和C=X基团(X为O、N、S、C),且与该基团相连的键上具有γ氢原子时,γ氢原子可重排(转移)到不饱和基团上(通常通过六元环过渡态),同时β键发生断裂,脱去一个中性分子,产生McLafferty重排。在醛、酮、酸、酯、酰胺、羰基衍生物、烯、炔及烷基苯等化合物的质谱中均可发现这种重排离子峰。
②RDA重排:RDA重排是以六元环烯化合物的双键为裂解反应起点,由π电子提供的游离基反应中心引发,通过两次α开裂,形成一个中性分子(乙烯或其衍生物)和一个离子化的共轭双烯衍生物。在环烯化合物、生物碱、萜类、甾体、黄酮及某些邻位二取代芳烃等化合物的质谱中,经常出现由RDA重排产生的碎片离子峰,可为上述化合物的结构分析提供重要信息。
(4)质谱解析
质谱解析的一般程序如下:确认分子离子峰,确定分子量;根据分子离子峰的丰度,推测化合物的可能类别;根据分子离子峰与同位素峰的丰度比,判断分子中是否含有高丰度的同位素元素,如Cl、Br、S 等,并推算这类元素的种类及数目;由同位素峰强比法或精密质量法确定分子式,并由分子式计算不饱和度,了解双键数及环数;分析基峰及主要碎片离子峰可能代表的结构单元,由此确定化合物可能含有的官能团,并参考其他光谱(波谱)数据,推测出所有可能的结构式;根据标准谱图及其他所有信息,进行筛选验证,确定化合物的结构式。
(5)综合波谱解析
以多种波谱数据对有机物结构进行综合解析时,要灵活应用各图谱提供的信息。综合波谱解析的一般程序如下:
①分子式的确定:目前分子式的确定主要有以下几种方法:a)元素分析法:采用元素分析仪定量测出分子中C、H、N、O、S等元素的含量,据此计算出各元素的原子比,拟定实验式,最后根据相对分子质量和实验式确定分子式;b)质谱法:由高分辨质谱获得化合物的精确相对分子质量,采用精密质量法确定分子式。若采用低分辨质谱,则可采用同位素相对强度法,由M+1、M+2与M峰的相对丰度比,并利用Beynon 表,确定化合物的分子式;c)核磁共振波谱法:核磁共振碳谱可提供化合物中碳原子数目,辅以氢谱,可方便地推算分子式。只有氢谱时,因峰面积与氢核数成正比,分子中氢核的总数将是这些峰面积最简比例总和的整数倍。如能得到化合物的相对分子质量、元素分析数据及NMR波谱数据,即可计算分子中的C原子数,从而确定分子式。
②结构单元确定:a)计算不饱和度:由分子式计算不饱和度,并结合各图谱,初步推断未知物的类别;b)利用各谱的特征信息初步确定结构单元:一般紫外光谱可判断有无共轭体系;红外光谱可判断化合物类别和有哪些基团存在,以及该基团与其他基团相连接的信息;NMR氢谱的偶合裂分及化学位移常常是推断相邻基团的重要线索, NMR碳谱的δ值以及是否表现出分子的对称性,对确定取代基的相互位置十分有用;质谱的主要碎片离子间的质量差值以及重要重排离子等,均可得出基团间相互连接的信息;c)结构单元的推断:一般先以一种图谱的信息为基础,推断可能属于哪一类化合物及结构单元,然后再以其他图谱加以验证。一般说来,对某一给定的结构单元,必须在各图谱中均能得到印证方可确认。若在某一图谱中未出现,则应重新考虑所推断结构的合理性或在哪个环节发生了错误。采用这种由少至多逐渐增加信息量的方式,可最有效地得出最终结论;d)考察剩余结构单元:从分子式(或相对分子质量)中扣除已确定的各结构单元的分子式(或相对分子质量),推测出剩余结构单元的不饱和度及可能结构。
③未知物结构的确定:a)以结构单元组成几种可能结构:若所确定结构单元中不饱和基团与分子的不饱和度相等,则可考虑它们之间各种连接顺序的可能性,并将确定的结构单元组成几种可能的分子结构;若所确定结构单元中不饱和基团的不饱和度低于分子的不饱和度,则在组成可能结构时还应考虑分子中环的组成;b)注意不饱和键及杂原子的位置:在组成分子的可能结构时,应注意安排好不饱和键及杂原子的位置(特别是杂原子的位置),因它们的位置对各谱均会产生重要的影响;c)推断最可能结构:以各图谱(多采用MS的裂解规律)对初步确定的几种结构进行核对。若所推测的某结构与已知图谱有明显矛盾时,说明该结构不合理,应删去;若所推测的几种结构均与各图谱大致相符时,说明推测的结构基本合理。再对某些碳原子或氢原子的δ值进行计算,从计算值与实测值相比的结果,以推断最可能的结构;d)确定最终结构:核对标准光谱或文献光谱,最终确定化合物的结构。
3.基本计算
(1)质谱方程式:
(2)质谱仪的分辨率:
(3)质量精度:质量精度=
(4)亚稳离子的质量:
第十六章色谱分析法概论 - 章节小结1.基本概念
保留时间t R:从进样到某组分在柱后出现浓度极大时的时间间隔。
死时间t0:分配系数为零的组分即不被固定相吸附或溶解的组分的保留时间。
调整保留时间t R':某组分由于溶解(或被吸附)于固定相,比不溶解(或不被吸附)的组分在柱中多停留的时间。
相对保留值r2,1:两组分的调整保留值之比。
分配系数K:在一定温度和压力下,达到分配平衡时,组分在固定相与流动相中的浓度之比。
保留因子k:在一定温度和压力下,达到分配平衡时,组分在固定相和流动相中的质量之比。
分离度R:相邻两组分色谱峰保留时间之差与两色谱峰峰宽均值之比。
分配色谱法:利用被分离组分在固定相或流动相中的溶解度差别或分配系数的差别而实现分离的色谱法。
吸附色谱法:利用被分离组分对固定相表面吸附中心吸附能力的差别或吸附系数的差别而实现分离的色谱法。
离子交换色谱法:利用被分离组分离子交换能力的差别或选择性系数的差别而实现分离的色谱法。
分子排阻色谱法:根据被分离组分分子的线团尺寸或渗透系数的差别而进行分离的色谱法。
涡流扩散:在填充色谱柱中,由于填料粒径大小不等,填充不均匀,使同一个组分的分子经过多个不同长度的途径流出色谱柱,使色谱峰展宽的现象。
纵向扩散:由于浓度梯度的存在,组分将向区带前、后扩散,造成区带展宽的现象。
传质阻抗:组分在溶解、扩散、转移的传质过程中所受到的阻力称为传质阻抗。
保留指数I:在气相色谱法中,常把组分的保留行为换算成相当于正构烷烃的保留行为,也就是以正构烷烃系列为组分相对保留值的标准,即用两个保留时间紧邻待测组分的基准物质来标定组分的保留,这个相对值称为保留指数,又称Kovats指数。
保留体积V R:是从进样开始到某组分在柱后出现浓度极大时,所需通过色谱柱的流动相体积。
调整保留体积V R':是由保留体积扣除死体积后的体积。
保留比R':设流动相的线速度为u,组分的移行速度为v,将二者之比称为保留比。
2.基本理论
(1)色谱分离的原理:组分在固定相和流动相间进行反复多次的“分配”,由于分配系数K(或容量因子k)的不同而实现分离。各种色谱法的分离机制不同。
(2)塔板理论:塔板理论描述组分在色谱柱中的分配和转移行为,由塔板理论导出的流出曲线方程为:
塔板理论有如下基本假设:①在色谱柱内一小段长度即一个塔板高度H内,组分可以在两相中瞬间达到分配平衡。②分配系数在各塔板上是常数。③试样和新鲜流动相都加在第0号塔板上。④流动相不是连续地而是间歇式地进入色谱柱,且每次只进入一个塔板体积。⑤试样在柱内的纵向扩散可以忽略。
塔板理论在解释流出曲线的形状和位置、组分的分离及评价柱效等方面是成功的。
(3)速率理论:速率理论解释了影响塔板高度或使色谱峰展宽的各种因素,包括涡流扩散、纵向扩散、传质阻抗和流动相线速度。其表达式为:H=A+B/u+Cu
A为涡流扩散系数:A=2ldp
B为纵向扩散系数:B=2gDm
C为传质阻抗:包括固定相传质阻抗Cs和流动相传质阻抗Cm
3.基本计算
(1)保留值:t R'=t R-t0,V R'=V R-V0,r2,1=t R1'/t R2'=V R1'/V R2'
(2)分配系数和保留因子:,,t R=t0(1+KVs/Vm) =t0(1+ k),k=t R'/t0(3)峰宽度:W1/2=2.355ζ,W=4ζ=1.699W1/2
(4)柱效:
(5)分离度:
二、重点和难点
本章主要学习色谱过程和分离原理、各类色谱的分离机制。尤其是色谱法的有关概念和色谱基本理论,是学习其后各章色谱分析方法的基础。
1.色谱过程
色谱过程是组分的分子在流动相和固定相间多次分配的过程。若两组分的分配系数存在微小的差异,经过反复多次的分配平衡,使微小的差异积累起来,其结果就使分配系数小的组分被先洗脱,从而使两组分得到分离。色谱分离的前提是分配系数或保留因子不等。
2.有关概念及计算公式
这是本章的重点,一定要深入理解,牢固掌握。
3.基本类型色谱方法及其分离机制
(1)分配色谱法:利用被分离组分在固定相或(和)流动相中的溶解度差别,即分配系数的差别而实现分离。包括气液分配色谱法和液液分配色谱法。
(2)吸附色谱法:利用被分离组分对固定相表面吸附中心吸附能力的差别,即吸附系数的差别而实现分离。包括气固吸附色谱法和液固吸附色谱法。
在硅胶液固吸附色谱中,极性强的组分吸附力强。常见化合物的吸附能力有下列顺序:烷烃<烯烃<卤代烃<醚<硝基化合物<叔胺<酯<酮<醛<酰胺<醇<酚<伯胺<羧酸。
(3)离子交换色谱法:利用被分离组分离子交换能力的差别即选择性系数的差别而实现分离。按可交换离子的电荷符号又可分为阳离子交换色谱法和阴离子交换色谱法。
(4)分子排阻色谱法:根据被分离组分分子的线团尺寸,即渗透系数的差别而进行分离。
分配色谱法是基础,而且在GC和HPLC中都还会有讨论。在TLC一章重点讨论吸附色谱法。后两种方法只存在于液相色谱法中,但在后续章中都没有专门讨论,故在本章加以介绍。
值得注意的是在实际色谱过程中各种分离机制极少单独发生,常常是几种机制同时发生,只是某种机制起主导作用而已。
4.塔板理论
塔板理论沿用分馏塔中塔板的概念来描述组分在两相间的分配行为。认为在每个塔板的间隔内,试样
组分在两相中达到分配平衡,经过多次的分配平衡后,分配系数小的组分先流出色谱柱。同时还引入塔板数作为衡量柱效的指标。而理论塔板数n可理解为在色谱柱内溶质平衡的次数(n=L/H),平衡的次数越多,
柱效越高,组分间分离的可能性越大。塔板理论实际上是把组分在两相间的连续转移过程,分解为间歇的在单个塔板中的分配平衡过程。
重点是要搞清溶质在色谱柱内的质量分配和转移。在色谱柱各塔板内组分的质量分布符合二项式
(m s+m m)N的展开式。
需要注意的是,在讨论二项式分布时,用二项式展开式或通式求得的Nmr是组分在色谱柱中各塔板内的溶质质量分数。当转移次数N=n(塔板数)时,柱出口开始能检测到溶质。流出曲线的纵坐标是柱出口处的质量分数,该曲线也符合二项式分布曲线。当塔板数很大时流出曲线趋于正态分布曲线。
5.速率理论
Van Deemter方程式为:H=A+B/u+Cu
速率理论的塔板高度H与塔板理论中的塔板高度有所不同,是色谱峰展宽的指标,但两者均是柱效的的度量。 B及C分别代表涡流扩散系数、纵向扩散系数和传质阻抗系数,其单位分别为cm、cm2/s及s。三者均与色谱动力学因素有关。重点是要理解这些影响柱效的因素的物理含义。
涡流扩散:也称为多径扩散,与填充不规则因子l和填料(固定相)颗粒的平均直径dp有关:A=2ldp 纵向扩散:纵向扩散系数B与弯曲因子g和组分在流动相中的扩散系数Dm有关:B=2gDm
传质阻抗:影响组分溶解、扩散、转移的阻力,包括固定相传质阻抗Csu和流动相传质阻抗Cmu。
流动相线速度对塔板高度的影响:在较低线速度时,纵向扩散项起主要作用,线速度升高,塔板高度降低,柱效升高;在较高线速度时,传质阻抗起主要作用,线速度升高,塔板高度增高,柱效降低。
速率理论研究影响柱效(或峰展宽即组分离散)的各种动力学因素,用于指导色谱实验条件的选择。Van Deemter方程在GC、HPLC和CE中的具体形式和应用将在相应章节讨论。根据此方程还可以求出流动相的最佳流速uop。以H=A+B/u+Cu对u微分,得H'=-Bu-2+C,当其等于0时,H有极值,于是-Bu-2+C=0,
因此,此时塔板高度为。
第十七章气相色谱法 - 章节小结1. 基本概念
固定液相对极性,麦氏常数,程序升温,噪声,漂移,分流比,检测器灵敏度,检测限等。
2.基本理论
(1)差速迁移:在色谱分析中,分配系数不同是组分分离的前提条件。气相色谱法中,载气种类少,可选余地小,要改变组分之间分配系数的或大小或比例,主要通过选择合适的固定液。
(2)GC中的速率理论:速率理论是从色谱动力学的角度阐述影响柱效的因素,以Van Deemter方程式表示,在填充柱中,速率方程为:
H=A+B/u+Cu =2λdp+ 2gDg/u+
在开管柱中,A=0,此时速率方程为:
H=B/u+Cgu+Clu =u +
最小板高对应的载气线速度称为最佳线速度,为了减少分析时间,常用的最佳实用线速度大于最佳线速度。在学习速率理论时,应熟悉速率方程式中各项和各符号的含义,即这些因素是如何影响柱效的,从而理解分离条件的选择。
(3)色谱柱分填充柱及毛细管柱两类,填充柱又分气-固色谱柱及气-液色谱柱。固定液按极性分类可分成非极性、中等极性、极性以及氢键型固定液。固定液的选择按相似性原则。常用硅藻土载体分为红色载体
和白色载体,红色载体常用于涂渍非极性固定液,白色载体常用于涂渍极性固定液。硅藻土载体常需进行钝化,其目的是为了减小载体表面的活性。载体钝化的方法有酸洗(AW)、碱洗(BW)和硅烷化,这些钝化方法分别除去碱性氧化物(主要是氧化铁)、酸性氧化物(氧化铝)和覆盖硅羟基。
毛细管柱可分为涂壁毛细管柱(WCOT)、载体涂层毛细管柱(SCOT)、多孔层毛细管柱(PLOT)和填充毛细管柱。
检测器分浓度型及质量型两类。氢焰检测器是质量型检测器,具有灵敏度高,检测限小,死体积小等优点。热导检测器是浓度型检测器,组分与载气的热导率有差别即能检测。电子捕获检测器也是一种浓度型检测器,检测含有强电负性基团的物质,具有高选择性和高灵敏度。
为保护检测器和色谱柱,开气相色谱仪时,必须先开载气,后开电源,加热。关机时,先关电源,最后关载气。
(4)柱温的选择原则为:在使最难分离的组分有尽可能好的分离度的前提下,要尽可能采用较低的柱温,但以保留时间适宜及不拖尾为度。对宽沸程样品,采用程序升温方式。
(5)定性与定量:定性方法有已知物对照法,相对保留值,保留指数,利用化学方法配合,两谱联用定性。定量方法常用归一化法和内标法,在没有校正因子情况下,使用内标对比法较好。
3.基本计算
固定液的相对极性
分离方程式 R=
相对重量校正因子=
归一化法 Ci%=
外标法 mi =
内标法 mi=fiAi ms=fsAs mi= Ci%=
内标对比法
第十八章高效液相色谱法 - 章节小结1.基本概念
(1)化学键合相:利用化学反应将有机基团键合在载体表面形成的固定相。
(2)化学键合相色谱法:以化学键合相为固定相的色谱法。
(3)正(反)相色谱法:流动相极性小(大)于固定相极性的液相色谱法。
(4)抑制型(双柱)离子色谱法:用抑制柱消除流动相的高电导本底,以电导为检测器的离子交换色谱法。
(5)手性色谱法:利用手性固定相或手性流动相添加剂分离分析手性化合物的对映异构体的色谱法。
(6)亲合色谱法:利用或模拟生物分子之间的专一性作用,从复杂生物试样中分离和分析特殊物质
的色谱方法,是基于组分与固定在载体上的配基之间的专一性亲和作用而实现分离的色谱法。
(7)梯度洗脱:在一个分析周期内程序控制改变流动相的组成,如溶剂的极性、离子强度和pH值等。
(8)静态流动相传质阻抗Csm:由于组分的部分分子进入滞留在固定相微孔内的静态流动相中,因
而相对晚回到流路中,引起的峰展宽。
(9)键合相的含碳量:键合相碳的百分数,可通过对键合硅胶进行元素分析测定。
(10)键合相的覆盖度:参加反应的硅醇基数目占硅胶表面硅醇基总数的比例。
(11)封尾:在键合反应后,用三甲基氯硅烷等对键合相进行钝化处理,减少残余硅醇基,即封尾。
(12)溶剂的极性参数P':表示溶剂与三种极性物质乙醇(质子给予体)、二氧六环(质子受体P')和硝基甲烷(强偶极体)相互作用的强度。用于度量分配色谱的溶剂强度。P'越大,溶剂的极性越强,在
正相分配色谱中的洗脱能力越强。
(13)溶剂的强度因子S:常为反相键合相色谱的溶剂洗脱能力的度量。
(14)三维光谱-色谱图:用DAD检测器检测,经过计算机处理,将每个组分的吸收光谱和试样的色谱图结合在一张三维坐标图上,即获得三维光谱-色谱图。
2.基本理论
(1)速率理论在HPLC中表达式为:H=A+C m u+C sm u 用于指导实验条件的选择。A、Cm和Csm均随固定相粒度dp变小而变小,因此保证HPLC高柱效的主要措施是使用小粒度的固定相。此外,要求粒度和柱填充
均匀、使用低粘度流动相、适当的流速和柱温。
(2)反相键合相色谱法保留机制:可用“疏溶剂理论”说明,即溶质的保留是其分子受到溶剂的斥力,而与键合相的烃基发生疏水缔合的结果。反相键合相色谱法的k受下列因素影响:①溶质的极性越强,k
越小;②流动相的含水量越高,使组分的k越大;③流动相的pH(离子抑制作用)和离子强度都影响k;
④键合相的烃基越长,使溶质k增大。
(3)正相键合相色谱法:以氨基、氰基等极性键合相为固定相,以烷烃加少量极性调节剂为流动相,极性强的组分k大。
(4)反相离子对色谱法:组分的离子与加入到流动相中的离子对试剂的反离子生成中性离子对,增加在反相固定相上的保留。保留因子受离子对试剂的种类和浓度、流动相的pH等影响。
3.基本计算
(1)混合溶剂的强度以下式计算:
式中P'为混合溶剂的极性参数,P i'和ji为纯溶剂i的极性参数及该溶剂在混合溶剂中的体积分数。
S混为混合溶剂的强度因子,Si和ji为纯溶剂i的强度因子及该溶剂在混合溶剂中的体积分数。
(2)HPLC的定量分析计算:与GC相同。
4.HPLC仪器
(1)输液泵
(2)检测器:有紫外、荧光、电化学和蒸发光散射检测器等。
紫外检测器:原理是朗伯-比尔(Lambert-Beer)定律;适用于有紫外吸收的组分。
二极管阵列检测器:可获得三维光谱-色谱图,同时提供定性定量信息。
荧光检测器:原理是荧光强度与组分的浓度成线性关系。
电化学检测器:原理是当组分经过电极表面时,发生氧化还原反应,产生电量(Q)的大小符合法拉第定律:Q=nFN;
蒸发光散射检测器:散射光的强度(I)与气溶胶中组分的质量(m)有下述关系: I=kmb或lgI=blgm+lgk 二、重点和难点
由于HPLC中的常用术语、色谱参数和色谱基本理论等都已在第16章中作了详细的阐述,定性、定量
分析方法又与GC相同,因此,本章只讨论高效液相色谱的各种具体方法,重点是反相键合相色谱法。
1.正相键合相色谱法
以极性键合相为固定相,如氰基(-CN)、氨基(-NH2)或二羟基键合相等,以非极性或弱极性溶剂,如烷烃,加适量极性调整剂,如醇类作流动相。正相键合相色谱主要用于分离溶于有机溶剂的中等极性的分子型化合物。分离选择性决定于键合相的种类、流动相的强度和试样的性质。主要是理解其一般规律:极性强的组分的保留因子k大,后洗脱出柱。流动相的极性增强,洗脱能力增加,使组分k减小,tR减小。
溶质与键合极性基团间的作用力,如氢键、诱导或静电作用等,是决定色谱保留和分离选择性的首要因素。流动相的选择性是通过其与试样分子间的相互作用来实现的,分子间作用力不同,则分离的选择性不同。同系物,如甲醇与丙醇,作用力类型相同,其色谱分离的选择性相似,如果换成高偶极矩溶剂二氯甲烷,则选择性将发生变化。
2.反相键合相色谱法
在现代色谱学中,反相键合相色谱法一般就称为反相色谱法。这是本章要掌握的重点。反相色谱法通常以非极性键合相为固定相,以极性溶剂及其混合物为流动相,固定相的极性比流动相的极性弱;能分离极性范围很宽的试样。键合相的烷基链长和含碳量是影响溶质的保留因子(及柱效)、选择性和载样量的重要因素。
保留行为的主要影响因素:①溶质的极性越弱,其疏水(疏溶剂)性越强,k越大,tR也越大。②增加流动相中水的含量,则溶剂强度降低,使溶质的k增大。③流动相的pH变化会改变溶质的离解程度,溶质的离解程度越高,k值越小。因此,常加入少量弱酸、弱碱或缓冲溶液,调节流动相的pH,抑制有机弱酸、弱碱的离解,增加它与固定相的疏水缔合作用,以达到分离的目的。这种色谱方法又称为离子抑制色谱法。④固定相键合烷基的碳链延长,硅胶表面键合烷基的浓度越大,溶质的k越大。
3.反相离子对色谱法
把离子对试剂加入到含水流动相中,与组分离子生成中性离子对,从而增加溶质与非极性固定相的作用,使分配系数增加,改善分离效果。用于分离可离子化或离子型的有机酸、碱、盐等。影响容量因子的因素有离子对试剂的种类和浓度、流动相的pH和流动相中有机溶剂的种类和比例等。
4.键合相的特点
键合相有如下优点:①使用过程中不流失;②化学稳定性好;③适于梯度洗脱;④载样量大。
使用一般化学键合相时流动相中水相的pH应维持在2~8,以免引起硅胶溶解,但目前已有许多键合相能够承受更宽的pH范围。
新型键合相
5.键合相色谱法中流动相对分离的影响根据分离方程式:
n由固定相及色谱柱填充质量决定,α(即选择性)主要受溶剂种类的影响,k受溶剂配比的影响。
溶剂的选择性:斯奈德(Snyder)根据溶剂的质子接受能力(Xe)、质子给予能力(Xd)和偶极作用力(Xn),将选择性参数定义为:
,,
根据Xe﹑Xd﹑Xn的相似性,将常用溶剂分为8组(教材表20-2),并得到溶剂选择性分类三角形。处于同一组中的各溶剂的作用力类型相同,在色谱分离中具有相似的选择性,而处于不同组别的溶剂,其选择性差别较大。
溶剂极性参数:
溶剂的极性和强度:溶剂的洗脱能力即溶剂强度直接与它的极性相关。在正相色谱中,溶剂极性参数P'值越大,则溶剂的极性越强,洗脱能力越强。
反相键合相色谱法的溶剂强度常用强度因子S表示。极性弱的溶剂S大,洗脱能力强。
混合溶剂的强度:
5.高效液相色谱中的速率理论
高效液相色谱的流动相是液体,液体与气体性质的差异是导致高效液相色谱的扩散和传质过程对柱效的影响与气相色谱有差别的主要原因。
(1)纵向扩散可忽略:纵向扩散系数β=2γDm,在HPLC中,液体流动相粘度大,使Dm很小。因此只有在流速很低时才能观察到纵向扩散对柱效的影响。在常用色谱条件下,纵向扩散可以忽略,故流速升高, H总是升高。但升高速率比GC中慢,因此,为了快速分析,一般仍选择大于最佳流速的条件。
(2)传质阻抗:化学键合相色谱法中,df可忽略,故固定相传质阻抗Cs可忽略。流动相传质阻抗Cm=ωmdp2/Dm中的ωm与柱内径、形状、填料性质等有关,但至今仍没确切的数值或关系。与GC中的Cs 不同,Cm与保留因子无关。HPLC中还有一项静态流动相传质阻抗Csm,而且Cm和Csm都与固定相的粒度dp有关。
(3)涡流扩散小:小颗粒球形固定相,匀浆高压填充,降低涡流扩散。
(4)HPLC中的范第姆特方程为:H=A+C m u+C sm u
(5)固定相粒度对塔板高度的影响:dp变小,A、Cm和Csm均变小,而且塔板高度受流动相线速度的影响也越小。可见小粒度的填料比在GC中更为重要。
根据速率理论,HPLC的实验条件应该是:①小粒度、均匀的球形化学键合相;②低粘度流动相,流速不宜快;③柱温适当。
6.反相键合相色谱实验条件的选择
在反相键合相色谱中,常选用非极性键合相。非极性键合相可用于分离分子型化合物,也可用于分离离子型或可离子化的化合物。ODS是应用最广泛的非极性固定相。对于各种类型的化合物都有很强的适应能力。短链烷基键合相能用于极性化合物的分离,苯基键合相适用于分离芳香化合物以及多羟基化合物如黄酮苷类等。
反相键合相色谱法中,流动相一般以极性最强的水为基础溶剂,加入甲醇、乙腈等极性调节剂。极性调节剂的性质以及其与水的混合比例对溶质的保留值和分离选择性有显著影响。一般情况下,甲醇-水已能满足多数试样的分离要求,且粘度小、价格低,是反相键合相色谱法最常用的流动相。乙腈的溶剂强度较高,且粘度较小,其截止波长(190nm)比甲醇(205nm)的短,更适合于利用末端吸收进行检测。
可选择弱酸(常用醋酸)、弱碱(常用氨水)或缓冲盐(常用磷酸盐及醋酸盐)作为抑制剂,调节流动相的pH,抑制组分的离解,增强保留。
调节流动相的离子强度也能改善分离效果,在流动相中加入0.1%~1%的醋酸盐、磷酸盐等,可减弱固定相表面残余硅醇基的干扰作用,减少峰的拖尾,改善分离效果。
第十九章平面色谱法- 章节小结
(一)平面色谱的基本术语和公式
(二)主要平面色谱类型
(三)吸附薄层色谱条件的选择
根据被测组分的极性大,选择吸附剂的活度要小,流动相极性要大;被测组分的极性小,选择吸附剂的活度要大,流动相极性要小。
1.被分离物质的极性与结构的关系
(1)基本母核相同,基团极性愈大,分子极性愈强;极性基团数目增加,分子极性增强。常见的取代基极性大小顺序:烷烃<烯烃<醚类<硝基化合物<二甲胺<脂类<酮类<醛类<硫醇<胺类<酰胺<醇类<酚类<羧酸类。
(2)分子双键愈多,共轭度愈大,吸附性愈大。
(3)空间排列影响极性,如能产生分子内氢键的分子极性下降。
2.吸附剂的活度选择被分离物质的极性大,吸附剂活度要小,以免吸附太牢,不易洗脱;被分离物质的极性小,则吸附剂的活度要大,以免不被吸附,而无法分离。可改变板活化温度和时间来控制吸附剂的活度。
3.展开剂的极性按相似相溶原则选择。
单一溶剂的极性顺序:石油醚<环己烷<二硫化碳<四氯化碳<三氯乙烷<苯、甲苯<二氯甲烷<氯仿<乙醚<乙酸乙酯<丙酮 <正丙醇<乙醇<甲醇<吡啶<酸<水。
4.混合溶剂选择的一般规则先用单一的中等极性展开剂试验,得出合适的极性。再改用二元展开剂,调节比例达到预期的极性,得到混合展开剂。目的是通过混合展开剂的极性和溶剂的强度,使各组分具有适宜的Rf值,从而达到良好的分离效率
分析化学(第7版)习题参考解答
第二章误差和分析数据处理 1、指出下列各种误差是系统误差还是偶然误差?如果是系统误差,请区别方法误 差、仪器和试剂误差或操作误差,并给出它们的减免方法。 答:①砝码受腐蚀: 系统误差(仪器误差);更换砝码。 ②天平的两臂不等长: 系统误差(仪器误差);校正仪器。 ③容量瓶与移液管未经校准: 系统误差(仪器误差);校正仪器。 ④在重量分析中,试样的非被测组分被共沉淀: 系统误差(方法误差);修正方法,严格沉淀条件。 ⑤试剂含被测组分: 系统误差(试剂误差);做空白实验。 ⑥试样在称量过程中吸潮: 系统误差(操作误差);严格按操作规程操作。 ⑦化学计量点不在指示剂的变色范围内: 系统误差(方法误差);另选指示剂。 ⑧读取滴定管读数时,最后一位数字估计不准: 偶然误差;严格按操作规程操作,增加测定次数。 ⑨在分光光度法测定中,波长指示器所示波长与实际波长不符: 系统误差(仪器误差);校正仪器。 ⑩在HPLC测定中,待测组分峰与相邻杂质峰部分重叠 系统误差(方法误差);改进分析方法 6、两人测定同一标准试样,各得一组数据的偏差如下: ②为什么两组数据计算出的平均偏差相等,而标准偏差不等; ③哪组数据的精密度高?
解:①n d d d d d 321n ++++= Λ 0.241=d 0.242=d 1 2 i -∑= n d s 0.281=s 0.312=s ②标准偏差能突出大偏差。 ③第一组数据精密度高。 7、测定碳的相对原子质量所得数据:12.0080、12.0095、12.0099、12.0101、12.0102、12.0106、12.0111、12.0113、12.0118及12.0120。求算:①平均值;②标准偏差;③平均值的标准偏差;④平均值在99%置信水平的置信限。 解:①12.0104i =∑= n x x ②0.00121)(2 i =--∑=n x x s ③0.00038== n s s ④0.0012 0.000383.25 25.3t 92-2 0.01±=?±==±±==时,,查表置信限=f n s t n s t x u 8、在用氯丁二烯氯化生产二氯丁二烯时,产品中总有少量的三氯丁二烯杂质存在。分析表明,杂质的平均含量为1.60%。改变反应条件进行试生产,取样测定,共取6次,测定杂质含量分别为:1.46%、1.62%、1.37%、1.71%、1.52%及1.40%。问改变反应条件后,产品中杂质百分含量与改变前相比,有明显差别吗?(α=0.05时) 解: %.n S S .S x x 05306/%13.0/%130 1.51% ===== 1.7% 053.0% 60.1%51.1=-= -= x S x t μ计 查表2-2,t 5,0.05=2.571,t 计算 < t 临界值 。 所以,改变反应条件后,杂志含量与改变前无明显差别。
1.下列四种硫的含氧酸盐中,氧化能力最强的是………………………………( )(A)Na2SO4(B)Na2S2O3(C)Na2S4O6(D)K2S2O8 2.下列物质中不是一元酸的是………………………………………………………………() (A) CH 3COOH (B) H 3 PO 3 (C) HNO 2 (D) H 3 PO 2 3.下列卤化物中,共价性最强的是……………………………………………………………() (A) BeI 2 (B) RbCl (C) LiI (D) LiF 4.下列离子中,最易水解的离子是……………………………………………………………() (A) Na+(B) Ca2+(C) Al3+(D) K+ 5. IVA族元素从Ge到Pb;下列性质随原子序数的增大而增加的是………………………() (A) +2氧化态的稳定性 (B) 二氧化物的酸性 (C) 单质的熔点(D) 氢化物的稳定性 6.铁的原子序数为 26,则Fe3+ 在强八面体场中的晶体场稳定化能(以△ =10 Dq表示)是…() (A) -20Dq(B) -20Dq+ 2P (C) -12Dq(D) -12Dq+ 2P 7.将K2MnO4溶液调节到酸性时,可以观察到的现象是………………………………………() (A)紫红色褪去(B)绿色加深 (C)有棕色沉淀生成(D)溶液变成紫红色且有棕色沉淀生成 8.在酸性介质中加入过氧化氢(H2O2)时不生成过氧化物的化合物是…………………………() (A) 钛酸盐(B) 重铬酸盐(C) 钒酸盐(D) 高锰酸盐
9. BF 3、B 2 H 6 、Al 2 Cl 6 都是稳定的化合物,BH 3 、AlCl 3 则相对不稳定,其原因是……… () (A) 前者形成大π键,后者缺电子 (B) 前者通过大π键、多中心键、配位键补偿了缺电子,后者缺电子 (C) 前者缺电子,后者有多中心键 (D) 前者有配位键,后者缺电子 10.[Ni(en) 3 ]2+离子中镍的价态和配位数是……………………………………………………() (A) +2,3 (B) +3,6 (C) +2,6 (D) +3,3 11.下列各组化合物中,不能稳定存在的一组物质是…………………………………………() (A) SiF 4,Si 3 N 4 (B) PbBr 4 ,PbI 4 (C) SnBr 4,SnI 4 (D) GeCl 2 ,PbF 4 12.在热碱性溶液中,次氯酸根离子不稳定,它的分解产物是………………………………() (A) Cl—(aq) 和Cl 2 (g) (B) Cl—(aq) 和(aq) (C) Cl—(aq) 和(aq) (D) Cl—(aq) 和(aq) 13.下列各对含氧酸盐热稳定性的大小顺序,正确的是………………………………………() (A) BaCO 3 > K 2 CO 3 (B) CaCO 3 < CdCO 3 (C) BeCO 3 > MgCO 3 (D) Na 2 SO 3 > NaHSO 3 14.干燥H 2 S气体,通常选用的干燥剂是………………………………………………………() (A) 浓H 2SO 4 (B) NaOH (C) P 2 O 5 (D) NaNO 3 15.某白色固体易溶于水,加入BaCl 2 有白色沉淀产生,用HCl酸化,沉淀完全溶解,再加入过量NaOH至强碱性,并加热,有刺激性气体逸出。此白色固体是………………………………()
第七章氧化还原滴定 1.条件电位和标准电位有什么不同?影响电位的外界因素有哪些? 答:标准电极电位E′是指在一定温度条件下(通常为25℃)半反应中各物质都处于标准状态,即离子、分子的浓度(严格讲应该是活度)都是1mol/l(或其比值为1)(如反应中有气体物质,则其分压等于1.013×105Pa,固体物质的活度为1)时相对于标准氢电极的电极电位。 电对的条件电极电位(E0f)是当半反应中氧化型和还原型的浓度都为1或浓度比为,并且溶液中其它组分的浓度都已确知时,该电对相对于标准氢电极电位(且校正了各种外界因素影响后的实际电极电位,它在条件不变时为一常数)。由上可知,显然条件电位是考虑了外界的各种影响,进行了校正。而标准电极电位则没有校正外界的各种外界的各种因素。 影响条件电位的外界因素有以下3个方面; (1)配位效应; (2)沉淀效应; (3)酸浓度。 2.是否平衡常数大的氧化还原反应就能应用于氧化还原中?为什么? 答:一般讲,两电对的标准电位大于0.4V(K>106),这样的氧化还原反应,可以用于滴定分析。 实际上,当外界条件(例如介质浓度变化、酸度等)改变时,电对的标准电位是要改变的,因此,只要能创造一个适当的外界条件,使两电对的电极电位超过0.4V ,那么这样的氧化还原反应也能应用于滴定分析。但是并不是平衡常数大的氧化还原反应都能应用于氧化还原滴定中。因为有的反应K虽然很大,但反应速度太慢,亦不符合滴定分析的要求。 3.影响氧化还原反应速率的主要因素有哪些? 答:影响氧化还原反应速度的主要因素有以下几个方面:1)反应物的浓度;2)温度;3)催化反应和诱导反应。 4.常用氧化还原滴定法有哪几类?这些方法的基本反应是什么? 答:1)高锰酸钾法.2MnO4+5H2O2+6H+==2Mn2++5O2↑+8H2O. MnO2+H2C2O4+2H+==Mn2++2CO2+2H2O 2) 重铬酸甲法. Cr2O72-+14H++Fe2+===2Cr3++Fe3++7H2O CH3OH+Cr2O72-+8H+===CO2↑+2Cr3++6H2O 3)碘量法3I2+6HO-===IO3-+3H2O, 2S2O32-+I2===2I-+2H2O Cr2O72-+6I-+14H+===3I2+3Cr3++7H2O 5.应用于氧化还原滴定法的反应具备什么条件? 答:应用于氧化还原滴定法的反应,必须具备以下几个主要条件: (1)反应平衡常数必须大于106,即△E>0.4V。 (2)反应迅速,且没有副反应发生,反应要完全,且有一定的计量关系。 (3)参加反应的物质必须具有氧化性和还原性或能与还原剂或氧化剂生成沉淀的物质。 (4)应有适当的指示剂确定终点。 6.化学计量点在滴定曲线上的位置与氧化剂和还原剂的电子转移数有什么关系? 答:氧化还原滴定曲线中突跃范围的长短和氧化剂与还原剂两电对的条件电位(或标准电位)相差的大小有关。电位差△E较大,突跃较长,一般讲,两个电对的条件电位或标准电位之差大于0.20V时,突跃范围才明显,才有可能进行滴定,△E值大于0.40V时,可选用氧化还原指示剂(当然也可以用电位法)指示滴定终点。 当氧化剂和还原剂两个半电池反应中,转移的电子数相等,即n1=n2时,则化学计量点的位
分析化学上册总答案
第2章 2、为了了解磷肥对水稻生长的影响,若果从某稻田中取一小铲泥样进行测定,试问由此试样所得分析结果有无意义?为什么? 答:无意义,这样的分析结果没有代表性。了解磷肥对水稻生长的影响因素需要对土壤进行分析,土壤的分析内容十分丰富,包括成分分析,肥力分析和污染物分析等。土壤的组成具有不均匀性,且影响因素十分复杂,为了采得具有代表性的试样,必须多方面考虑。比如:采样点的布设,采样时间,采样深度,采样量等。 5、已知铝锌矿的K=0.1,a=2。 (1)采取的原始试样最大颗粒直径为30mm,问最少应采取多少千克试样才具有代表性?(2)将原始试样破碎并通过直径为3.36mm的筛孔,再用四分法进行缩分,最多应缩分几次?(3)如果要求最后所得分析试样不超过100克,问试样通过筛孔的直径应为多少毫米? 解:(1)K=0.1 a=2 m =Kd a=0.1×(30)2=0.1×900=90kg (2) K=0.1 a=2 m= Kd a=0.1×(3.36)2=1.13kg (90/2n)<1.13 n=6.33 故最少缩分6次。 (3) Kd2≧0.1 d=1mm 第4章 2.指出下列情况下,各会引起哪种误差?如果是系统误差,应该采取什么方法减免? (1)电子天平未经校准;(2)容量瓶何以业管部配套; (3)试剂中含有微量的被测组分;(4)天平的零点有微小波动; (5)滴定时从锥形瓶中溅出一滴溶液; 。 (6)标定HCl溶液用的NaOH标准溶液中吸收了CO 2 答:(1)系统误差中的仪器误差。减免的方法:校准仪器或更换仪器。 (2)系统误差中的仪器误差。减免的方法:校准仪器或更换仪器。 (3)系统误差中的试剂误差。减免的方法:做空白实验。 (4)随机误差。(5)过失误差。 (6)系统误差中的试剂误差。减免的方法:做空白实验。 8.下列数据中包括极为有效数字? =4.74 (6)pH=10.00 (1)0.0083 (2)27.160 (3)700.0 (4)7.80 (5) pK a 答:(1)2 (2)5 (3)4 (4)3 (5)2 (6)2 10两位分析者同时测定某一合金中铬的质量分数,每次称取试样均为2.00g,分别报告结果如下:甲:1.02%,1.03%;乙:1.018%,1.024%。问哪一份报告是和合理的,为什么?
分析化学期末复习资料 第一章 绪论 1. 分析化学包括:定量分析,定性分析,结构分析(形态分析) 2. 定性分析的对象包括:样本,分析物 3. 按样本大小可分为:常量分析(固:>100mg ,液:>10mL ),半微量分析(固:10~100mg ,液:1~10mL ),微量分析(固:0.1~10mg ,液:0.01~1mL ) 4. 按分析物在样品中所占含量可分为:主要(常量)组分分析(>1%),微量组分分析(0.01~1%),痕量组分分析(<0.01%) 第二章 误差及分析数据的统计处理 1. 误差:测定值与真值之间的差值。 2. 绝对误差:E=T X - 3. 相对误差:Er= ?-T T X 100% 4. 准确度:测定平均值与真值接近的程度,常用误差大小表示。误差小,准确度高。 5. 偏差:个别测定结果与几次测定结果的平均值之间的差值。 6. 绝对偏差:di =Xi-X 7. 相对偏差:dr = ?-X X Xi 100% 8. 相对平均偏差:??-= ∑X n X Xi dr 100% 9. (样本)标准偏差:s= () 1 -n X -Xi n 1 i 2 ∑= 10. 精密度:在相同条件下,多次重复测定值相互符合的程度,常用偏差大小表 示。 11. 实验结果首先要求精密度高,才能保证有准确的结果,但高的精密度不一定能保证有高的准确度。(如无系统误差存在,则精密度高,准确度也高) 12. 校正系统误差(准确度,误差)的方法:改进方法,校正仪器,对照实验,空白实验,回收率实验 13. 校正随机(偶然)误差(精密度,偏差)的方法:增加测定次数 14. 随机误差大,系统误差不一定大 15. 可疑值的取舍:G 检验(书p17)(G= s X X i -,s :标准偏差),Q 检验(p18) 16. 平均值的置信区间(p14):n s t X ?± =μ(t :查表可得,n ;测定次数)
1.下列数据中有效数字不是四位的是( ) (A)0.2500 (B)0.0025 (C) 2.005 (D)20.50 2. 用分析天平准确称取0.2g 试样,正确的记录应是( ) (A)0.2g (B)0.20g (C) 0.200g (D)0.2000g 3. 欲配制pH=5的缓冲溶液,下列物质的共轭酸碱对中最好选择( ) A. 一氯乙酸(K a = 1.4×10-3) B. 甲酸(K a = 1.8×10-4) C. 乙酸(K a = 1.8×10-5) D. 氨水(K b = 1.8×10-5) 4. 在pH= 5.0的醋酸缓冲液中用0.02 mol·L -1的 EDTA 滴定同浓度的Pb 2+。已知:lg K PbY =18.0,lg Y(H) =6.6,lg Pb(Ac) =2.0,化学计量点时溶液中pPb’应为( ) A. 10. B. 9.0 C. 6.7 D. 5.7 5. 某HCl 溶液中c Fe(III)=c Fe(Ⅱ)=1mol ?L -1,则此溶液中铁电对的条件电位'θ?为( ) A. ' θFe /Fe 23+ + ?= θFe /Fe 23++? B. ' θFe /Fe 23++?= θFe /Fe 23++?) () (II Fe III Fe lg 059.0αα+ C. ' θFe /Fe 23++?= θFe /Fe 23++?) () (II Fe Fe III Fe Fe 32lg 059.0αγαγ+ + + D. ' θFe /Fe 23++?= θFe /Fe 23++?) () (III Fe Fe II Fe Fe 23lg 059.0αγαγ+ + + 6.反应 2A ++ 3B 4+ → 2A 4++3B 2+到达化学计量点时电位是( ) A. 2 θB θA ??+ B. 5 6 )2(θ B θA ?+?? C. 5 32θ B θA ??+ D. 523θB θA ??+ 7. 在下列各组酸碱组分中,不属于共轭酸碱对的是( ) A. HOAc-NaOAc B. H 3PO 4-H 2PO 4- C. +NH 3CH 2COOH-NH 2CH 2COO - D. H 2CO 3-HCO 3- 8. 莫尔法测定Cl -含量时,要求介质的pH6.5~10.0范围内,若pH 过高,则
2008 –2009 学年第二学期期末考试分析化学试卷 一、选择题(每题2分,共30分) 1.定量分析结果的标准偏差代表的是-----------------------------(C )。 A. 分析结果的准确度 B. 分析结果的精密度和准确度 C. 分析结果的精密度 D. 平均值的绝对误差 2.下列哪种情况应采用返滴定法-------------------------------------(C )。 A. 用AgNO3标准溶液测定NaCl试样含量 B. 用HCl标准溶液测定Na2CO3试样含量 C. 用EDTA标准溶液测定Al3+试样含量 D. 用Na2S2O3标准溶液测定K2Cr2O7试样含量 3.下列各项叙述中不是滴定分析对化学反应要求的是----------(D )。 A. 反应必须有确定的化学计量关系 B. 反应必须完全 C. 反应速度要快 D. 反应物的摩尔质量要大 4. 下列四个数据中为四位有效数字的-------------------------------- ( C ) (1)0.0056 (2)0.5600 (3)0.5006 (4)0.0506 A. 1, 2 B. 3, 4 C. 2, 3 D. 1, 4 5. 以下有关随机误差的论述正确的是------------------------------( C ) A. 正误差出现概率大于负误差 B. 负误差出现概率大于正误差 C. 正负误差出现的概率相等 D. 大小误差出现的概率相等 6. 在用K2Cr2O7法测定Fe 时, 加入H3PO4的主要目的是--( B ) A. 提高酸度, 使滴定反应趋于完全 B. 降低化学计量点前Fe3+/Fe2+电对的电位,使二苯胺磺酸钠在突跃范围内变 色 C. 提高化学计量点前Fe3+/Fe2+电对的电位, 使二苯胺磺酸钠不致提前变色 D. 有利于形成Hg2Cl2白色丝状沉淀 7. 用Fe3+滴定Sn2+在化学计量点的电位是--------------------------( D ) [ ?' (Sn4+/Sn2+)=0.14V] ?' (Fe3+/Fe2+)=0.68V, A. 0.75V B. 0.68V C. 0.41V D. 0.32V 8. 测定试样中CaO 的质量分数, 称取试样0.9080 g,滴定耗去EDTA 标准溶液20.50 mL, 以下结果表示正确的是---------------( C ) A. 10% B. 10.1% C. 10.08% D. 10.077% 9. 下列滴定分析操作中会产生系统误差的是-----------------------( C ) A. 指示剂选择不当 B. 试样溶解不完全 C. 所用蒸馏水质量不高 D. 称样时天平平衡点有±0.1mg的波动 10. 某溶液含Ca2+、Mg2+及少量Al3+、Fe3+,今加入三乙醇胺, 调至pH=10, 以铬 黑T为指示剂, 用EDTA滴定, 此时测定的是---------( A )
第一章概论 1—3 分析全过程: 取样、处理与分解;试样的分离与富集;分析方法的选择;结果的计算与评价. 1-4 标定碱标准溶液时,邻苯二甲酸氢钾(KHC8H4O4, M=204。23g.mol—1)和二水合草酸(H2C2O4. 2H2O, M=126。07g.mol—1)都可以作为基准物质,你认为选择哪一种更好?为什么? 答:选择邻苯二甲酸氢钾更好。因为邻苯二甲酸氢钾的摩尔质量较大,称量误差较小。 1—5.基准物Na2CO3和Na2B4O7·10H2O都可用于标定HCl溶液的浓度.你认为选择哪一种更好为什么答:选择Na2B4O7·10H2O更好.因为Na2B4O7·10H2O的摩尔质量较大,称量误差较小 1—6 用基准Na2CO3标定HCl溶液时,下列情况会对HCl的的浓度产生何种影响(偏高、偏低或没有影响)? a。滴定时速度太快,附在滴定管壁的HCl来不及流下来就读取滴定体积 b. 称取Na2CO3时,实际质量为0.0834g,记录时误记为0。1824g c. 在将HCl标准溶液倒入滴定管之前,没有用HCl溶液荡洗滴定管 d。锥瓶中的Na2CO3用蒸馏水溶解时,多加了50mL蒸馏水 e。滴定开始之前,忘记调节零点,HCl溶液的液面高于零点 f。滴定管活塞漏出HCl溶液 g. 称取Na2CO3时,撇在天平盘上 h. 配制HCl溶液时没有混匀 答:使用Na2CO3标定HCl的浓度时,HCl的浓度计算公式为:c HCl=2m Na2CO3/(M Na2CO3V HCl)。 a。由于V HCl偏高,c HCl偏低; b. 由于m Na2CO3偏低,c HCl偏低; c. 由于V HCl偏高,c HCl偏低; d. 无影响; e。因为V HCl偏低,c HCl偏高; f. 因为V HCl偏高,c HCl偏低; g. 由于Na2CO3易吸湿,应用减量法称量。称取Na2CO3时,在天平盘上,Na2CO3会吸湿,使m Na2CO3偏低,最终导致c HCl偏低; h. 溶液没有混匀时,很可能的情况是上层较稀,因此c HCl偏低的可能性较大. 1—7. 若将H2C2O4·2H2O基准物质不密封,长期置于放有干燥剂的干燥器中,用它标定NaOH溶液的浓度时,结果是偏高,偏低,还是无影响 答:若将未密封H2C2O4·2H2O基准物质长期置于放有干燥剂的干燥器中,会使其失去结晶水。用它标定NaOH溶液的浓度时,消耗NaOH溶液的体积偏高。根据,最终使结果偏低。 1—8. 假设用HCl标准溶液滴定不纯的Na2CO3试样,若出现7题中所述的情况,将会对分析结果产生何
- 242O C 电位法及永停滴定法 1.在25℃,将pH 玻璃电极与饱和甘汞电极浸入pH=6.87的标准缓冲溶液中,测得电动势为0.386V ;测定另一未知试液时,测得电动势为0.508V 。计算未知试液的pH 。 94.8059 .0386 .0508.06.87 059.0=-+=-+=S X S X E E pH pH 2. 若K H+,Na+=1×10-15,这意味着提供相同电位时,溶液中允许Na +浓度是H +浓度的多少倍?若Na +浓度为1.0 molL 时,pH=1 3.00的溶液所引起的相对误差是多少? (1)K H+,Na+=1×10-15时,意味着干扰离子Na +的活度比被测离子H +的活度高1×1015倍时,两者才产生相同的电位。 (2)%0.110 .10016.0101%100%13 15=??=??=?--x n n Y Y X a a K C C Y X , 3.某钙离子选择电极的选择系数K Ca2+,Na+=0.0016,测定溶液中Ca 2+离子的浓度,测得浓度值为 2.8×10-4molL ,若溶液中存在有0.15molL 的NaCI ,计算:①由于NaCl 的存在,产生的相对误差是多少?②若要使相对误差减少到2%以下,NaCl 的浓度不能大于多少? ()%89.12%100108.215.00016.0%100%4 1 /2=???= ??=?-x n Y Y X a a K C C Y X , 若要使相对误差减少到2%以下,则 ()%100108.20016.0%100%24 1 /2???= ??>-x x n n Y Y X C a a K Y X , 解得NaCl 的浓度不能大于0.059molL 4.用下列电池按直接电位法测定草酸根离子浓度。 Ag │AgCl (固)│KCl (饱和┊┊ (未知浓度)│Ag 2C 2O 4(固)│Ag (1)推导出pC 2O 4与电池电动势之间的关系式(Ag 2C 2O 4的溶度积K sp =2.95×10- 11) (2)若将一未知浓度的草酸钠溶液置入此电解池,在25℃测得电池电动势为0.402V ,Ag-AgCl 电极为负极。计算未知溶液的pC 2O 4值。 (已知A g C l /A g ?= + 0.1990V ,Ag /Ag +?=+0.7995V ) 4 24211////2059 .04889.02059 .0)1095.2lg(2059.07995.0lg 2059.0lg 2059.0lg 059.0lg 059.024 224 24 22O pC O pC K K O C sp Ag Ag O C sp Ag Ag Ag Ag Ag Ag O C Ag +=+?+=-+=+=+=--+- +++α?α?α??θθ θθ 4 242////2 059 .02899.01990.02059.04889.0lg 059.0422O pC O pC E Ag Ag Ag Ag Ag Ag Ag Ag O C Ag +=-+=-+=-=++++?α???θ 80 .32 059 .02899.0402.04242=+=O pC O pC
分析化学在现实生活中的应用我们的生活离不开物质。如何让物质能更加美好我们的生活呢?掌握一点化学知识其实是非常实用的方法。无论是生产、生活,还是环境保护、能源与资源的利用、医药卫生与人体健康等与化学有着广泛的关系。因此,生活中有许多化 学知识需要我们去认识。 “民以食为天”,我们先来看看吃里的化学吧。 油条是我国传统的早餐食品之一,它的历史非常悠久。当大家吃着香脆可口的油条时,是否会想到油条制作过程中的化学知识呢? 先来看看油条的制作过程:首先是发面,用鲜酵母或老面(酵面)与面粉一起加水揉和,使面团发酵到一定程度后,再加入适量纯碱、食盐和明矾进行揉和,然后切成厚1厘米,长10厘米左右的条状物,把每两条上下叠好,用窄木条在中间压一下,旋转后拉长放入热油锅里去炸,便成了一根香、脆的油条。 在发酵过程中,由于酵母菌在面团里繁殖分泌酵素(主要是分糖化酶和酒化酶),使一小部分淀粉变成葡萄糖,又由葡萄糖变成乙醇,并产生二氧化碳气体。同时,还会产生一些有机酸类,这些有机酸与乙醇作用生成有香味的酯类。反应产生的二氧化碳气体使面团产生许多小孔并且膨胀起来。有机酸的存在,就会使面团有酸味,加入纯碱,就是要把多余的有机酸中和掉,并能产生二氧化碳气体,使面团进一步膨胀起来;同时,纯碱溶于水发生水解,后经热油锅一炸,由于有二氧化碳生成,使炸出的油条更加疏松。 从上面的反应中,也许大家会担心,在制作油条时不是使用了氢氧化钠吗?含有如此强碱的油条,吃起来怎么会可口呢?然而其巧奥妙之处也在于此。当面团里出现游离的氢氧化钠时,原料中的明矾就立即跟它发生了反应,使游离的氢氧化钠经成了氢氧化铝。氢氧化铝的凝胶液或干燥凝胶,在医疗上用作抗酸药,能中和胃酸、保护溃疡面,用于治疗胃酸过多症、胃溃疡和十二指肠溃疡等。常见的治
智慧树知到《分析化学》章节测试答案 第一章 1、某组分的百分含量为97.5%,其有效数字位数是()。 A:一位 B:两位 C:三位 D:四位 正确答案:四位 2、下列情况引起偶然误差的是( )。 A:重量法测定SiO2时,硅酸沉淀不完全 B:使用腐蚀了的砝码进行称量 C:滴定管读数最后一位估计不准 D:所有试剂中含有干扰组分 正确答案:滴定管读数最后一位估计不准 3、提高分析结果准确度的方法有() A:选择恰当的分析方法 B:减小测量误差 C:减小偶然误差的影响 D:消除测量中的系统误差 正确答案:选择恰当的分析方法,减小测量误差,减小偶然误差的影响5、置信度愈高,置信区间愈大。 A:对
B:错 正确答案:错 第二章 1、下列各组酸碱对中属于共轭酸碱对的是() A:H2CO3~CO3- B:H3O+~OH- C:H3PO4~H2PO4- D:H2SO4~SO4- 正确答案:H3PO4~H2PO4- 2、可用于标定硫代硫酸钠的基准物质有()。 A:A.KIO3 B:B.KBrO3 C:C.K2Cr2O7 D:D.K3[Fe(CN)] 正确答案: A.KIO3,C.K2Cr2O7 3、下列溶液以NaOH溶液或盐酸溶液滴定时,在滴定曲线上会出现二个突跃的是()。A:H2SO4+ H3PO4 B:HCl+ H3BO3 C:HAc+HF D:NaOH+ Na3PO4 正确答案:H2SO4+ H3PO4,NaOH+ Na3PO4 4、下列质子条件正确的是()。
A:NaCN溶液的质子条件是[H+]=[OH-]+[HCN] B:B.浓度为c的Na2S溶液的质子条件是[OH-]=[H+]+[HS-]+2[H2S] C:浓度为c的(NH4)2CO3溶液的质子条件是[H+]=[OH-]+[NH3]-[H CO3-] D:浓度为c的NaOH溶液的质子条件是[OH-]=[H+]+c 正确答案: NaCN溶液的质子条件是[H+]=[OH-]+[HCN],B.浓度为c的Na2S溶液的质子条件是[OH-]=[H+]+[HS-]+2[H2S],浓度为c的(NH4)2CO3溶液的质子条件是[H+]=[OH-]+[NH3]-[H CO3-],浓度为c的NaOH溶液的质子条件是[OH-]=[H+]+c 5、液体试样保存期的长短与待测物的稳定性以及保存方法有关。 A:对 B:错 正确答案:对 第三章 1、测量次数较多时,用标准偏差来表示一组数据的精密度 A:对 B:错 正确答案:A 2、酸碱滴定中选择指示剂的原则是() A:指示剂变色范围与化学计量点完全符合 B:指示剂应在pH=7.00时变色 C:指示剂的变色范围应全部或部分落入滴定pH突跃范围之内 D:指示剂的变色范围应全部落在滴定pH突跃范围之内 正确答案:C
第一章绪论 1、仪器分析和化学分析: 仪器分析是以物质的物理性质和物理化学性质(光、电、热、磁等)为基础的分析方法,这类方法一般需要特殊的仪器,又称为仪器分析法;、 化学分析是以物质化学反应为基础的分析方法。 2、标准曲线与线性范围: 标准曲线是被测物质的浓度或含量与仪器响应信号的关系曲线; 标准曲线的直线部分所对应的被测物质浓度(或含量)的范围称为该方法的线性范围。 3、灵敏度、精密度、准确度和检出限: 物质单位浓度或单位质量的变化引起响应信号值变化的程度,称为方法的灵敏度; 精密度是指使用同一方法,对同一试样进行多次测定所得测定结果的一致程度; 试样含量的测定值与试样含量的真实值(或标准值)相符合的程度称为准确度; 某一方法在给定的置信水平上可以检出被测物质的最小浓度或最小质量,称为这种方法对该物质的检出限。 第三章光学分析法导论 1、原子光谱和分子光谱: 由原子的外层电子能级跃迁产生的光谱称为原子光谱; 由分子的各能级跃迁产生的光谱称为分子光谱。 2、原子发射光谱和原子吸收光谱: 当原子受到外界能量(如热能、电能等)的作用时,激发到较高能级上处于激发态。但激发态的原子很不稳定,一般约在10-8 s内返回到基态或较低能态而发射出的特征谱线形成的光谱称为原子发射光谱; 当基态原子蒸气选择性地吸收一定频率的光辐射后跃迁到较高能态,这种选择性地吸收产生的原子特征的光谱称为原子吸收光谱。 3、线光谱和带光谱: 4、光谱项和光谱支项; 用n、L、S、J四个量子数来表示的能量状态称为光谱项,符号为n 2S + 1 L; 把J值不同的光谱项称为光谱支项,表示为n 2S + 1 L J。 5、统计权重和简并度; 由能级简并引起的概率权重称为统计权重; 在磁场作用下,同一光谱支项会分裂成2J+1个不同的支能级,2J+1称为能级的简并度。 6、禁戒跃迁和亚稳态; 不符合光谱选择定则的跃迁叫禁戒跃迁; 若两光谱项之间为禁戒跃迁,处于较高能级的原子具有较长的寿命,原子的这种状态称为亚稳态。 7、 8、 9、分子荧光、磷光和化学发光; 荧光和磷光都是光致发光,是物质的基态分子吸收一定波长范围的光辐射激发至单重激发态,再由激发态回到基态而产生的二次辐射。 荧光是由单重激发态向基态跃迁产生的光辐射, 磷光是单重激发态先过渡到三重激发态,再由三重激发态向基态跃迁而产生的光辐射。 化学发光是化学反应物或反应产物受反应释放的化学能激发而产生的光辐射。 10、拉曼光谱。 拉曼光谱是入射光子与溶液中试样分子间的非弹性碰撞引起能量交换而产生的与入射光频率不同的散射光形成的光谱。 第四章原子发射光谱法
第二章误差和分析数据处理 1、指出下列各种误差是系统误差还是偶然误差?如果是系 统误差,请区别方法误差、仪器和试剂误差或操作误差,并给出它们的减免方法。 答:①砝码受腐蚀: 系统误差(仪器误差);更换砝码。 ②天平的两臂不等长: 系统误差(仪器误差);校正仪器。③容量瓶与移液管未经校准:系统误差(仪器误差);校正仪器。④在重量分析中,试样的非被测组分被共沉淀:系统误差 (方法误差);修正方法,严格沉淀条件。⑤试剂含被测组分: 系统误差(试剂误差);做空白实验。⑥试样在称量过程中吸潮: 系统误差(操作误差);严格按操作规程操作。⑦化学计量点不在指示剂的变色范围内:系统误差(方法误差);另选指示剂。⑧读取滴定管读数时,最后一位数字估计不准:偶然误差;严格按操作规程操作,增加测定次数。⑨在分光光度法测定中,波长指示器所示波长与实际波长不符: 系统误差(仪器误差);校正仪器。 ⑩在HPLC 测定中,待测组分峰与相邻杂质峰部分重叠 系统误差(方法误差);改进分析方法
6、两人测定同一标准试样,各得一组数据的偏差如下: (1) 0.3 -0.2 -0.4 0.2 0.1 0.4 0.0 -0.3 0.2 -0.3 (2) 0.1 0.1 -0.6 0.2 -0.1 -0.2 0.5 -0.2 0.3 0.1 ① 求两组数据的平均偏差和标准偏差; ② 为什么两组数据计算出的平均偏差相等,而标准偏差不等; ③ 哪组数据的精密度高? ②标准偏差能突出大偏差。 ③ 第一组数据精密度高 7、测定碳的相对原子质量所得数据: 12.0080、12.0095、 12.0099、 12.0101、12.0102、12.0106、12.0111、12.0113、12.0118 及 12.0120。 求算:①平均值;②标准偏差;③平均值的标准偏差;④平均值在 99%置信水平的置信限。 解:① x x i 12.0104 n 2 ② s (x i x) 0.0012 n1 解: d 1 d 2 d 3 d n d 1 0.24 d 2 0.24 s 1 0.28 s 2 0.31 d s
分析化学期末考试试卷 班级:_______________学号:_______________:_______________得 分:_______________ (卷面共有44题,总分100分,各大题标有题量和总分,每小题标号后有小分) 一、选择(16小题,共26分) (1)称取钢样2.000 g,充分燃烧后产生的SO2通入50.00 mol 0.01000 mol/L NaOH溶液中吸收,过量的NaOH用0.01000 mol/L HCl溶液返滴定至酚酞终点,消耗30.00 mL,则钢样中硫的质量分数为 ( ) [A r(S)=32.06, H2SO3的p K a1=1.89,p K a2=7.20] (A) 0.16 (B) 0.32 (C) 0.08 (D) 0.64 (2)以下物质必须采用间接法配制标准溶液的是( ) (A) K2Cr2O7 (B) Na2S2O3 (C) Zn (D) H2C2O4·2H2O (3)使用碱式滴定管进行滴定的正确操作是 ( ) A 用左手捏稍低于玻璃珠的近旁 B用左手捏稍高于玻璃珠的近旁 C 用左手捏玻璃珠上面的橡皮管 D用右手捏稍低于玻璃珠的近旁 (4)用基准邻苯二甲酸氢钾标定NaOH溶液时,下列情况对标定结果产生负误差的是 ( ) A 标定完成后,最终读数时,发现滴定管挂水珠 B 规定溶解邻苯二甲酸氢钾的蒸馏水为50ml,实际用量约为60ml
C 最终读数时,终点颜色偏深 D 锥形瓶中有少量去离子水,使邻苯二甲酸氢钾稀释 (5) HPO42-的共轭碱是 ( ) (A) H2PO4- (B) H3PO4 (C) PO43- (D) OH- (6)用NaOH标准溶液滴定0.1mol/LHCl-0.1mol/L H3PO4混合液,在滴定曲线上出现几个突跃 ( ) (A) 1 (B) 2 (C) 3 (D) 4 (7)下列溶液用酸碱滴定法能准确滴定的是 ( ) (A) 0.1 mol/L HF (p K a = 3.18) (B) 0.1 mol/L HCN (p K a = 9.21) (C) 0.1 mol/L NaAc [p K a(HAc) = 4.74] (D) 0.1 mol/L NH4Cl [p K b(NH3) = 4.75] [1分](8)EDTA的酸效应曲线是指 ( ) (A) αY(H)-pH 曲线 (B) pM-pH 曲线 (C) lg K'(MY)-pH 曲线 (D) lgαY(H)-pH 曲线 (9)用EDTA直接滴定有色金属离子,终点所呈现的颜色是 ( ) (A) 指示剂-金属离子络合物的颜色 (B) 游离指示剂的颜色
分析化学期末试题 班级 学号 姓名 一、单项选择题(15分,每小题1分) 1、在以EDTA 为滴定剂的络合滴定中,都能降低主反应能力的一组副反应系数为( A )。 A 、αY(H), αY(N),αM(L); B 、αY(H), αY(N),αMY ; C 、αY(N), αM(L),αMY ; D 、αY(H),αM(L),αMY 。 2、在EDTA 络合滴定中,使滴定突跃增大的一组因素是( B )。 A 、C M 大,αY(H)小,αM(L)大,K MY 小; B 、C M 大,αM(L)小,K MY 大,αY(H) 小; C 、C M 大,αY(H)大, K MY 小,αM(L)小; D 、αY(H)小,αM(L)大,K MY 大,C M 小; 3、以EDTA 为滴定剂,下列叙述错误的是( D )。 A 、在酸度较高的溶液中,可形成MHY 络合物。 B 、在碱性较高的溶液中,可形成MOHY 络合物。 C 、不论形成MHY 或MOHY ,滴定反应进行的程度都将增大。 D 、不论溶液pH 值的大小,只形成MY 一种形式络合物。 4、在络合滴定中,有时出现指示剂的“封闭”现象,其原因为( D )。 (M :待测离子;N :干扰离子;In :指示剂) A 、''NY MY K K >; B 、' 'NY MY K K <; C 、''MY MIn K K >; D 、''MY NIn K K >。 5、在用EDTA 测定Ca 2+、Mg 2+的含量时,消除少量Fe 3+、Al 3+干扰的下述方法中,哪一种是正确的( C )。 A 、于pH=10的氨性缓冲溶液中直接加入三乙醇胺;
第一章概论 1-3 分析全过程: 取样、处理与分解;试样的分离与富集;分析方法的选择;结果的计算与评价。 1-4 标定碱标准溶液时,邻苯二甲酸氢钾(KHC8H4O4, M= 2H2O, M=,你认为选择哪一种更好?为什么? 答:选择邻苯二甲酸氢钾更好。因为邻苯二甲酸氢钾的摩尔质量较大,称量误差较小。 1-5.基准物Na2CO3和Na2B4O7·10H2O都可用于标定HCl溶液的浓度.你认为选择哪一种更好为什么 答:选择Na2B4O7·10H2O更好.因为Na2B4O7·10H2O的摩尔质量较大,称量误差较小 1-6 用基准Na2CO3标定HCl溶液时,下列情况会对HCl的的浓度产生何种影响(偏高、偏低或没有影响)? a. 滴定时速度太快,附在滴定管壁的HCl来不及流下来就读取滴定体积 b. 称取Na2CO3时,实际质量为0.0834g,记录时误记为0.1824g c. 在将HCl标准溶液倒入滴定管之前,没有用HCl溶液荡洗滴定管 d. 锥瓶中的Na2CO3用蒸馏水溶解时,多加了50mL蒸馏水 e. 滴定开始之前,忘记调节零点,HCl溶液的液面高于零点 f. 滴定管活塞漏出HCl溶液 g. 称取Na2CO3时,撇在天平盘上 h. 配制HCl溶液时没有混匀 答:使用Na2CO3标定HCl的浓度时,HCl的浓度计算公式为:c HCl=2m Na2CO3/(M Na2CO3V HCl)。 a. 由于V HCl偏高,c HCl偏低; b. 由于m Na2CO3偏低,c HCl偏低; c. 由于V HCl偏高,c HCl偏低; d. 无影响; e. 因为V HCl偏低,c HCl偏高; f. 因为V HCl偏高,c HCl偏低; g. 由于Na2CO3易吸湿,应用减量法称量。称取Na2CO3时,在天平盘上,Na2CO3会吸湿,使m Na2CO3偏低,最终导致c HCl偏低; h. 溶液没有混匀时,很可能的情况是上层较稀,因此c HCl偏低的可能性较大。 1-7. 若将H2C2O4·2H2O基准物质不密封,长期置于放有干燥剂的干燥器中,用它标定NaOH溶液的浓度时,结果是偏高,偏低,还是无影响 答: 若将未密封H2C2O4·2H2O基准物质长期置于放有干燥剂的干燥器中,会使其失去结晶水.用它标定NaOH 溶液的浓度时,消耗NaOH溶液的体积偏高.根据,最终使结果偏低. 1-8. 假设用HCl标准溶液滴定不纯的Na2CO3试样,若出现7题中所述的情况,将会对分析结果产生何种影响
第二章 误差和分析数据处理 1、 指出下列各种误差是系统误差还是偶然误差?如果是系统误差,请区别方法误差、仪器和试剂误差或操作误差,并给出它们的减免方法。 答:①砝码受腐蚀:系统误差(仪器误差);更换砝码。 ②天平的两臂不等长:系统误差(仪器误差);校正仪器。 ③容量瓶与移液管未经校准:系统误差(仪器误差);校正仪器。 ④在重量分析中,试样的非被测组分被共沉淀:系统误差(方法误差);修正方法,严格沉淀条件。 ⑤试剂含被测组分:系统误差(试剂误差);做空白实验。 ⑥试样在称量过程中吸潮:系统误差(操作误差);严格按操作规程操作。 ⑦化学计量点不在指示剂的变色范围内:系统误差(方法误差);另选指示剂。 ⑧读取滴定管读数时,最后一位数字估计不准:偶然误差;严格按操作规程操作,增加测定次数。 ⑨在分光光度法测定中,波长指示器所示波长与实际波长不符:系统误差(仪器误差);校正仪器。 ⑩在HPLC 测定中,待测组分峰与相邻杂质峰部分重叠:系统误差(方法误差);改进分析方法 6、两人测定同一标准试样,各得一组数据的偏差如下: ② 为什么两组数据计算出的平均偏差相等,而标准偏差不等; ③ 哪组数据的精密度高? 解:①n d d d d d 321n ++++= 0.241=d 0.242=d 1 2i -∑=n d s 0.281=s 0.312=s ②标准偏差能突出大偏差。 ③第一组数据精密度高。 7、测定碳的相对原子质量所得数据:12.0080、12.0095、12.0099、12.0101、12.0102、12.0106、12.0111、12.0113、12.0118及12.0120。求算:①平均值;②标准偏差;③平均值的标准偏差;④平均值在99%置信水平的置信限。 解:①12.0104i =∑= n x x ②0.00121)(2 i =--∑=n x x s ③0.00038==n s s ④0.0012 0.000383.25 25.3t 92-2 0.01±=?±==±±==时,,查表置信限=f n s t n s t x u 8、在用氯丁二烯氯化生产二氯丁二烯时,产品中总有少量的三氯丁二烯杂质存在。分析表明,杂质的平均含量为1.60%。改变反应条件进行试生产,取样测定,共取6次,测定杂质含量分别为:1.46%、1.62%、1.37%、1.71%、1.52%及1.40%。问改变反应条件后,产品中杂质百分含量与改变前相比,有明显差别吗?(α=0.05时) 解: %.n S S .S x x 05306/%13.0/%130 1.51%===== 1.7%053.0%60.1%51.1=-=-= x S x t μ 计 查表2-2,t 5,0.05=2.571,t 计算 < t 临界值 。 所以,改变反应条件后,杂志含量与改变前无明显差别。 9、解:HPLC 数据 : 97.2%,98.1%,99.9%,99.3%,97.2%,98.1%(6次) %1.1 %3.98==S x , 化学法数据: