
第21卷第4期V ol 121N o 14 三明高等专科学校学报JOURNA L OF S ANMI NG C O LLEGE
2004年12月
Dec 12004
收稿日期:2004206202
作者简介:黄 晖(1967-),女,福建闽清人,三明学院化生工程系高级讲师。
浊点萃取技术在痕量金属离子测定中的应用
黄 晖1,陈培珍2
(1.三明学院化生工程系,福建三明 365004;
2.南平师范高等专科学校化学与环境工程系,福建南平 353000)
摘 要:介绍了绿色分析法———浊点萃取技术及其在痕量金属离子分析测定中的应用。关键词:浊点萃取;表面活性剂;痕量金属离子;应用
中图分类号:O658.2 文献标识码:A 文章编号:1671-1343(2004)04-0066-06
浊点萃取技术(Cloud P oint Extraction ,CPE )是近年来出现的一种新兴的环保型液—液萃取技术,它具有经济、安全、高效、操作简便且应用范围广等优点,已成功地应用于生命科学和环境科学的研究中。对环境样品中痕量金属离子测定,其关键在于样品中的痕量金属离子的分离和富集,过去我们常常使用有机溶剂对金属离子和显色剂形成的金属螯合物进行萃取分离,从而达到分离和预富集痕量金属离子的目的,但该过程需要使用大量的易挥发的有毒有机溶剂,它不仅耗费试剂、污染环境,而且还影响分析操作人员的身体健康。而新兴的浊点萃取技术它以表面活性剂的浊点现象为基础,通过改变实验参数(如温度等)引发相分离,作为测定环境样品中痕量金属离子的前处理手段,在分离和富集痕量金属离子方面起着重要作用。它不使用挥发性的有机溶剂,不影响环境,表面活性剂用量小,萃取分离速度快,且萃取富集率最高可达100%,是高灵敏度、高选择性的“绿色分析方法”。
1 表面活性剂胶束溶液的形成和浊点现象
表面活性剂分子通常由疏水基和亲水基两部分组成。疏水部分在水溶液中聚集成核,亲水部分向外张开形成胶束。表面活性剂的重要功能有二,其一是它的增容作用(S olubiliza 2tion )[1]。其二是浊点(Cloud P oint ,CP )现象[2]。在一定的温度范围内,表面活性剂易溶于水成
为澄清的溶液,而当温度升高(或降低)到一定程度时,表面活性剂在水中的溶解度反而减小,会在水溶液中出现混浊、析出或分层的现象。这种混浊溶液静置一段时间(或离心)后形成透明的两液相,一相为量少(一般为100~200μL )且富含被萃取物的表面活性剂相,另一相为大量且表面活性剂胶束浓度为临界胶束浓度的水相。上述过程是可逆的,当温度向着相反方向变化时,即可恢复为均相溶液。
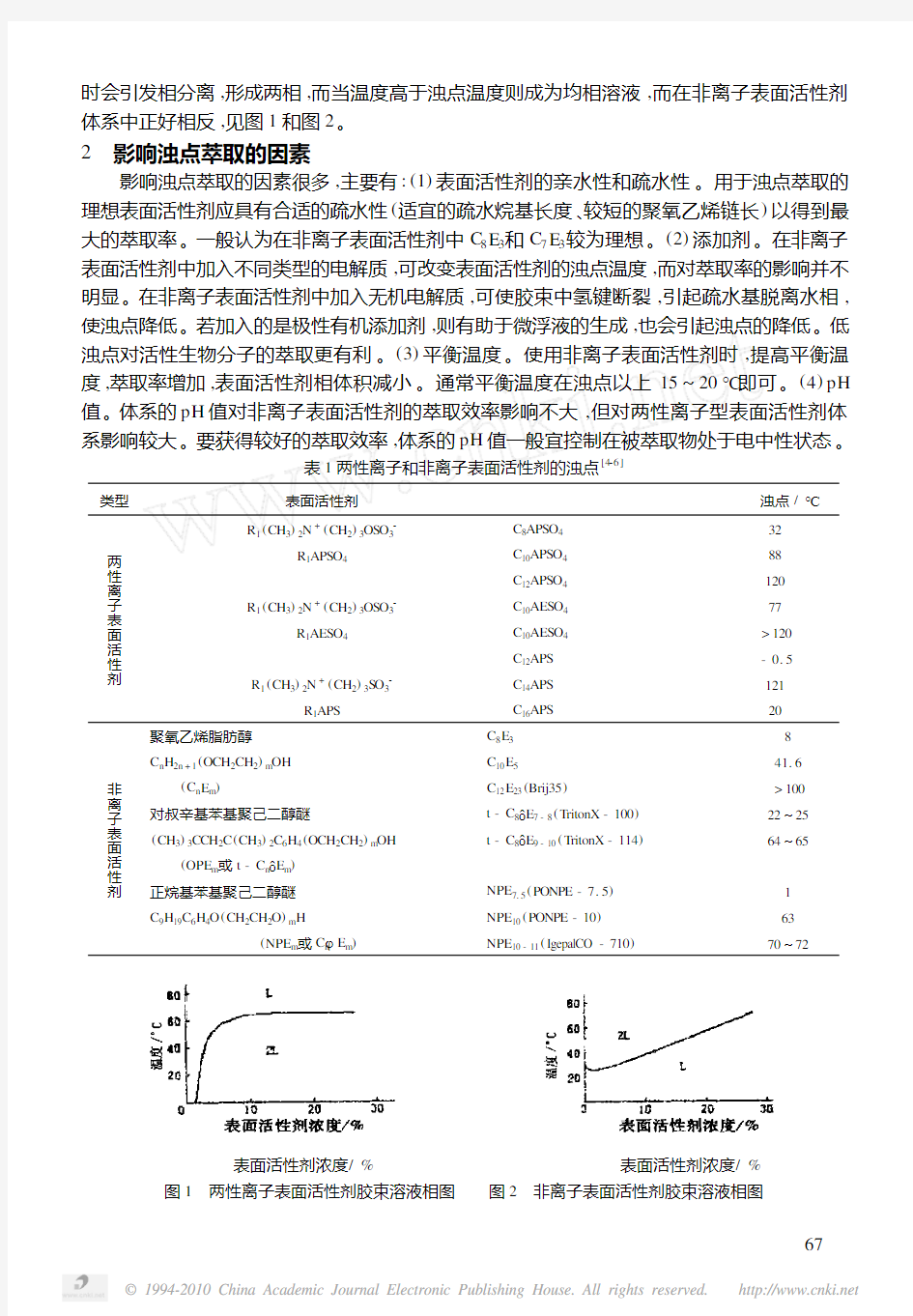
表面活性剂的相分离一般与表面活性剂的类型有关[3]。通常把表面活性剂分为:两性离子表面活性剂(包括阳、阴离子表面活性剂)和非离子表面活性剂。典型的两性离子和非离子表面活性剂类型及它们的浊点见表1。在两性离子表面活性剂体系中,当温度低于浊点温度
时会引发相分离,形成两相,而当温度高于浊点温度则成为均相溶液,而在非离子表面活性剂体系中正好相反,见图1和图2。
2 影响浊点萃取的因素
影响浊点萃取的因素很多,主要有:(1)表面活性剂的亲水性和疏水性。用于浊点萃取的理想表面活性剂应具有合适的疏水性(适宜的疏水烷基长度、较短的聚氧乙烯链长)以得到最大的萃取率。一般认为在非离子表面活性剂中C 8E 3和C 7E 3较为理想。(2)添加剂。在非离子表面活性剂中加入不同类型的电解质,可改变表面活性剂的浊点温度,而对萃取率的影响并不明显。在非离子表面活性剂中加入无机电解质,可使胶束中氢键断裂,引起疏水基脱离水相,使浊点降低。若加入的是极性有机添加剂,则有助于微浮液的生成,也会引起浊点的降低。低浊点对活性生物分子的萃取更有利。(3)平衡温度。使用非离子表面活性剂时,提高平衡温度,萃取率增加,表面活性剂相体积减小。通常平衡温度在浊点以上15~20℃即可。(4)pH 值。体系的pH 值对非离子表面活性剂的萃取效率影响不大,但对两性离子型表面活性剂体系影响较大。要获得较好的萃取效率,体系的pH 值一般宜控制在被萃取物处于电中性状态。
表1两性离子和非离子表面活性剂的浊点[426]
类型
表面活性剂
浊点/℃
两
性离子表面活性剂
R 1(CH 3)2N +(CH 2)3OS O 32
R 1APS O 4
R 1(CH 3)2N +(CH 2)3OS O 32
R 1AES O 4
R 1(CH 3)2N +(CH 2)3S O 32
R 1APS
C 8APS O 432C 10APS O 488C 12APS O 4
120C 10AES O 477C 10AES O 4>120C 12APS -0.5C 14APS 121C 16APS 20非离子表面活性剂
聚氧乙烯脂肪醇
C 8E 38C n H 2n +1(OCH 2CH 2)m OH
C 10E 5
41.6 (C n E m )
C 12E 23(Brij35)
>100对叔辛基苯基聚己二醇醚
t -C 8 E 7-8(T ritonX -100)22~25(CH 3)3CCH 2C (CH 3)2C 6H 4(OCH 2CH 2)m OH
t -C 8 E 9-10(T ritonX -114)64~65
(OPE m 或t -C n E
m )正烷基苯基聚己二醇醚
NPE 7.5(PONPE -7.5)1C 9H 19C 6H 4O (CH 2CH 2O )m H
NPE 10(PONPE -10)63(NPE m 或C n φE m )
NPE 10-11(IgepalCO -710)
70~72
表面活性剂浓度/% 表面活性剂浓度/%图1 两性离子表面活性剂胶束溶液相图 图2 非离子表面活性剂胶束溶液相图
以往虽有一些灵敏度较高的痕量金属离子的测定方法报道,但仍然不能满足环境样品中痕量金属离子的测定要求,环境样品中痕量金属离子的准确测定仍需要对环境样品中痕量金属离子进行浓缩和预富集,液-液萃取为最常用的富集方法,但液-液萃取操作麻烦,溶剂消耗量大,对环境有污染,也影响操作人员的身体健康,且引入的误差因素较多。浊点萃取技术最早是由Hiroto Watanabe and Hiroaki T anaka[7]应用于金属离子的测定的,随着浊点萃取技术的发展,尤其在生物大分子分离分析中取得的突破性进展,为浊点萃取在痕量金属离子测定中的进一步完善奠定了基础。通过表面活性剂对环境样品中痕量金属离子进行分离和预富集,再结合火焰原子吸收分光光度法(FAAS)、石墨炉原子吸收分光光度法(G FAAS)、等离子发射光谱(ICP)、毛细管电泳(CE)及紫外、可见分光光度法(UV/VIS)等分析方法或与流动注射分析技术(FI A)联用,实现了环境样品中痕量金属离子的准确测定。其萃取、富集过程如图3,该方面的相关研究见表2。
表2 浊点萃取技术在测定痕量金属离子的应用实例
金属离子螯合剂表面活性剂检测仪器样品文献
C o PAN,PAR,
52Br2PADAP
T ritonX2100
FAAS
ET AAS
Pharmaceutical
sam ple
[8]
Ag Dithiz one T ritonX2114FAAS W ater sam ple[9]
Al PONPE7.5ICP2OES2FI Parenteral
s olutions
[10]
Cr S DS T ritonX2114C L2FI W ater sam ple[11]
V52Br2PADAP PONPE5.0ICP2OES2FI Parenteral
s olutions
[12\〗
Hg52Br2PADAP PONPE5.0ICP2OES2FI T ap water[13] Hg Dithiz one T ritonX2100SRC2FI Natural water[14] Hg52Br2PADAP PONPE5.0ICP W ater[15] Hg52Br2PADAP PONPE5.0ICP W ater[16]
Cu DDTP T ritonX2100FAAS W ater,Serum,
Human hair
[17]
Cu OP210AAS Natural water[18] Cu DDTP T ritonX2100FAAS水样[19] Pb PONPE7.5FAAS Human saliva[20] C o,Ni T AN T ritonX2114FAAS W ater[21] Cd,Pb DDTP T ritonX2114FAAS Human hair[22]
Ru,Rh,Pd Pt,Au DDTP T ritonX2114ET V2ICP2MS
Biological
sam ples
[23]
Ag,As,Au,Cd
Cu,Pb,Se
DDTP T ritonX2114FI2ICP2MS W ater sam ples[24\〗Cd,Cu,Pb,Zn T AN T ritonX2114FAAS W ater sam ples[25] Cr,Zn,Cd,Pb APDC X2100:X245(6:4)FAAS W ines[26]
Cd,Pb,Cr,Cu Zn,Ni,Fe APDC T ritonX2114
FAAS
ICP2AES
Environmental
sam ples
[27]
4 浊点萃技术目前尚存在的问题
不可否认,浊点萃取技术作为新兴的样品前处理手段,在其理论上的研究还不完善,如表面活性剂产生浊点现象的原因还不十分清楚,非离子表面活性剂的相分离理论至今也尚无定论。其次,浊点萃取技术在痕量金属离子的萃取富集方面的研究,国外进行的相对较多,国内在该方面的研究相对薄弱。再者,浊点萃取技术与其它分析仪器联用的研究有待深入,如与高效液相色谱(HP LC)、毛细管电泳(CE)的联用技术,而且,由于有胶束的存在,浊点萃取特别适用于与胶束电动液相色谱(MEK C)的联用。
图3 典型的浊点萃取过程图解
Ⅰ、加入螯合剂、非离子表面活性剂,进行pH调节后,加热Ⅱ、离心分相Ⅲ、弃去水相
①金属离子 ②表面活性剂胶束 ③金属螯合物 ④富含表面活性剂相 ⑤水相
5 结论及展望
浊点萃取技术在萃取和富集痕量金属离子方面有以下突出的优点:(1)安全低毒,表面活性剂使用后可回收或焚化,对环境不造成污染。(2)萃取率高,富集因子大,最高萃取率可达100%。(3)萃取速度快,耗时短,操作方便。(4)表面活性剂用量小,成本低廉。(5)易与其它分析仪器联用,如与流动注射仪取用,可实现样品分离、富集和测定的自动化。(6)可实现大批量连续分析。总之,随着浊点萃取技术研究的不断成熟和完善,在痕量金属离子的分析测试中具有良好的发展前景。
参考文献:
[1]梁治齐,李金华.功能性浮化剂与浮化液[M].北京:中国轻工业出版社,2000.
[2]侯万国,孙德军,张春光.应用胶体化学[M].北京:科学出版社,1998.
[3]I.Casero,D.sicilia,S.Rubio,and D.perez2Bendito.An Acid2Induced Phase Cloud Point Separation Approach Using
Anionic Sur factants for the Extraction and Preconcentration o f Organic Compounds[J].Anal.Chem.,1999(71):45192 4526.
[4]Willie L Hinze,E Pramauro.A Critical Review o f Sur factant2Mediated Phase Separations(Cloud Point Extractions):
Theory and Application[J].Crit.Rev.Anal.Chem.,1993(24):1332177.
[5]Raym ond P.Frankewich and Willie L.Hinze.Evaluation and Optimization o f the Factor s Affecting Nonionic Sur factant2
Mediated Phase Separations[J].Anal.Chem.,1994(66):9442954.
[6]Frank H Quina.Sur factant2Mediated Cloud Point Extractions:An Environmentally Benign Alternative Separation Ap2
proach[J].Ind.Eng.Chem.Res.,1999(38):415024168.
[7]Hiroto Watanabe and Hiroaki T anaka.A Non2ionic Sur factant as a New Solvent for Liquidliquid Extraction o f Zinc(II)
with12(22pyridylazo)222naphthol[J].Talanta,1978(25):5852589.
[8]Clesia C.Nascentes,Marco Aurelio Z.Arruda.Cloud Point Formation Based on Micelles in the Presence o f Electrolytes
for Cobalt Extraction and Preconcentration[J].Talanta,2003(61):7592768.
[9]Jamshid L.Manzoori,G hasem K arim2Nezhad.Selective Cloud Point Extraction and Preconcentration o f Trace Amounts o f
Silver as a Dithizone Complex prior to Flame Atomic Absorption Spectrometric Determination[J].Analytica Chemica Acta, 2003(484):1552161.
[10]Lorena L.S ombra,Marta O.Luconi,Liliana P.Fernandez,et al.Assessment o f Trace Aluminium Content in Par2
enteral Solutions by Combined Cloud Point Preconcentration2flow Injection Inductively Coupled Plasma Optical Emission Spectrometry[J].Journal o f Pharmaceutical and Biomedical Analysis,2003(30):145121458.
[11]Exangelos K.Paleolog os,Athanasios G.Vlessidis,M iltiades I.K arayannis,et al..On2line Sorption Preconcentration
o f Metals Based on Mixed Micelle Cloud Point Extraction prior to Their Determination with Micellar Chemiluminescence Application to the Determination o f Chromium at ng L21levels[J].Analytica Chemica Acta,2003(477):2232231. [12]G ustav o M.Wuilloud,Jorgelina C.A.de Wuilloud,R odolfo G.Wuilloud,et al.Cloud Point Extraction o f Vanadi2
um in Parenteral Solutions Using a Nonionic Sur factant(PON PE5.0)and Determination by Flow injection2Inductively Coupled Plasma Optical Emission Spectrometry[J].Talanta,2002(58):6192627.
[13]Jorgelina C.A.de Wuilloud,R odolfo G.Wuilloud,Maria F.S ilva,et al.Sensitive Determination o f Mercury in Tap
Water by Cloud Point Extraction Pre2concentration and Flow injection2cold Vapor2inductively Coupled Plasma Optical E2 mission Spectrometry[J].Spectrochimmca Acta Part B,2002(57):3652374.
[14]M.G arrido,M.S.Di Nezio,A.G.Lista,et al.Cloud Point Extraction/Preconcentration on2line Flow Injection
Method for Mercury Determination[J].Analytica Chemica Acta,2004(502):1732177.
[15]S.Mc Intosh.The Determination o f Mercury at Ultratrace Levels Using and Automated Amalgamation Technique[J].At.
Spectrosc,1993,14(2):47249.
[16]V.S tresko,J.P olakovikova,J.K ubova.Atomic Absorption Spectrometric Determination o f Ultratrace Amounts o f Mer2
cury in Water[J].At.Spectrosc,1994(9):117321175.
[17]Jamshid L.Manzoori,Ahad Bavili2T abrizi.The Application o f Cloud Point Preconcentration for the Determination o f Cu
in Real Samples by Flame Atomic Absorption Spectrometry[J].Microchemical Journal,2002(72):127.
[18]S.A.K ulichenko,V.O.D oroschuk,S.O.Lelyushok.The Cloud Point Extraction o f Cu(Ⅱ)with Monocarboxylic
Acids into Non—ionic Sur factant Phase[J].Talanta,2003(59):7672773.
[19]陈建荣,林建军.浊点萃取—火焰原子吸收光谱法测定水样中痕量铜的研究[J].分析试验室,2002,21
(5):86289.
[20]Marta O.Luconi,M.Fernanda S ilva,R oberto A.Olsina,et al.Cloud Point Extraction o f Lead in Saliva via Use o f
Nonionic PON PE7.5without Added Chelating Agents[J].Talanta,2000(51):1232129.
[21]Jianrong Chen,K hay Chuan T eo.Determination o f Cobalt and Nickel in Water Samples by Flame Atomic Absorption
Spectrometry after Cloud Point Extraction[J].Analytica Chemica Acta,2001(434):3252330.
[22]Jamshid L.Manzoori,Ahad Bavili2T abrizi.Cloud Point Preconcentration and Flame Atomic Absorption Spectrometry
Determination o f Cd and Pb in Human hair[J].Analytica Chemica Acta,2002(470):2152221.
[23]Marcia Andreia Mesquita da S ilva,Vera Lucia Azzolin Frescura,Adils on Jose Curtius.Determination o f Noble Metals
in Biological Samples by Electrothermal Vaporization Inductively Coupled Plasma Mass Spectrometry,Following Cloud
Point Extraction[J].Spectrochimica Acta Part B,2001(56):194121949.
[24]Marcia Andreia Mesquita da S ilva,Vera Lucia Azzolin Frescura,Adils on Jose Curtius.Determination o f Trace Elements
in Water Samples by Ultrasonic Nebulization Inductively Coupled Plasma Mass Spectrometry after Cloud Point Extraction [J].Spectrochimica Acta Part B,2000(55):8032813.
[25]Jianrong Chen,K hay Chuan T eo.Determination o f Cadmium,Copper,Lead and Zinc in Water Samples by Flame
Atomic Absorption Spectrometry after Cloud Point Extraction[J].Analytica Chemica Acta,2001(450):2152222. [26]E.K.Paleolog os,D.L.G iokas,S.M.T zouwara2K arayaimi,et al.Micelle Mediated Methodology for the Determi2
nation o f Free and Bound Iron in Wines by Flame Atomic Absorption Spectrometry[J].Analytica Chemica Acta,2002 (458):2412248.
[27]D.L.G iokas,L.P.Eksperiandova,A.B.Blank,et https://www.doczj.com/doc/5b8926103.html,parison and Evaluation o f Cloud Point Extraction and
Low2temperature Directed Crystallization as Preconcentration Tools for the Determination o f Trace Elements in Environmen2 tal Samples[J].Analytica Chemica Acta,2003(224347):129.
[责任编辑:赖文忠]
The Application of Cloud Point Extraction on
the Determination of T race Metal Ions
H UANG Hui1,CHE N Pei2zhen2
(1.Department o f Chemistry and Biology Engineering,Sanming College,Sanming365004,China;
2.Department o f Chemistry and Environmental Engineering,Nanping Teacher s College,Nanping353000,China)
Abstract:Cloud point extraction(CPE)was introduced as a green analytical method.In this paper the application of this technique for the determination of trace metal ions was reviewed.
K ey w ords:cloud point extraction(CPE);sur factant;trace metal ions;application
铝及铝合金化学分析方法 EDTA滴定法 第32部分:铋含量的测定 Na 2 编制说明(征求意见稿) 一、工作简况 1、任务来源及计划要求 根据国标委《国家标准委关于下达2018年第三批国家标准制修订计划的通知》(国标委综合〔2018〕60号)文件精神,《铝及铝合金化学分析方法第32部分:铋量的测定方法二:Na2EDTA滴定法》由全国有色金属标准化技术委员会负责归口,由广东省工业分析检测中心负责,项目计划编号为20182000-T-610,完成时间为2020年。 2018年3月14日~3月16日,全国有色金属标准化技术委员会于云南省昆明市组织召开有色金属标准工作会议,会议对国家标准《铝及铝合金化学分析方法第32部分:铋量的测定方法二:Na2EDTA滴定法》进行任务落实,由广东省工业分析检测中心负责起草,参与起草单位有长沙矿冶研究院有限责任公司、贵州省分析测试研究院、中国铝业郑州有色金属研究院有限公司、山东南山铝业股份有限公司、深圳市中金岭南有色金属股份有限公司、北矿检测技术有限公司、有研亿金新材料有限公司。 2、调研和分析工作的情况 在当前国家“一带一路”、“中国制造2025”、国际产能和装备制造合作等战略发展形势下,随着国内外铁路、航空、电力和核发展等有力推动,促使轻量化结构材料---铝合金的需求量不断增长。 现有的GB/T20975系列《铝及铝合金化学分析方法》中没有铋的检测方法,而现有的涉及铝及铝合金中铋元素的检测方法是YS/T 807.9-2012 《铝中间合金化学分析方法第9部分:铋含量的测定碘化钾分光光度法》,测定范围为1.00 %~11.00 %,相较于分光光度法,化学滴定法更适用于常量铋的测定,因此有必要制定铝及铝合金中铋的化学滴定法检测标准,Na2EDTA滴定法用于测定范围为2.50 %~12.00 %的铋量。 Na2EDTA滴定法测定结果具有准确度高、操作简便的特点。对铝及铝合金中铋的Na2EDTA 滴定法测定条件和测定方法进行系统研究,并确定方法的准确度及精密度,最终形成国家标准。方法测定范围为2.50 %~12.00 %。 3、起草单位情况 广东省工业分析检测中心是我国南方从事金属材料、冶金产品、化工产品、再生资源质量检测、欧盟环保(RoHS)指令的有害物质检测、金属材料综合利用检测与咨询、评价以及分析测试技术研究的专业机构。先后隶属于广州有色金属研究院、广东省工业技术研究院(广州有色金属研究院),2015年12月经广东省机构编制委员会批准成为广东省科学院属下的独立事业法人单位。中心是一个检测设备配套齐全、检测技术完备、人员结构合理、管理科学的检测机构。近十年来获得省部级科技进步奖20项。累计申请专利15件,其中授权发明专利5件、授权实用新型专利2件。承担国家、省级各类项目50余项,主持和参与国家、行业标准300余项,发表专著5部,发表论文300余篇。 4、主要工作过程 根据任务落实会议精神,我中心成立《铝及铝合金化学分析方法》起草课题小组,明确了标准的进度安排、任务分工、确定了编制标准的工作计划及技术路线,完成相应的方法研究工作,完成标准相关工作。 (1)2018年3月14日~3月16日在云南省昆明市组织召开有色金属标准工作会议。对《铝及铝合金化学分析方法第32部分:铋量的测定方法二:Na2EDTA滴定法》标准进行了任务落实,批准了由广东省工业分析检测中心负责起草,长沙矿冶研究院有限责任公司、
离子注入材料表面改性的研究方法 【摘要】本文论述了离子注入材料表面改性的特点和发展应用,阐述了离子注入材料表面改性的机理。大量研究表明,离子注入通过改变材料表面和界面的物理化学特性及微观结构,能够显著提高材料的抗磨损,抗疲劳,抗腐蚀,抗氧化特性。离子注入不仅可以提高材料表面性能,延长材料使用寿命,还可以节约贵金属资源,具有很好的经济效益和应用前景。 【关键词】离子注入技术;材料表面改性;研究方法 1.前言 20世纪70年代,离子注入应用于材料表面改性并逐渐发展成一种新颖有效的材料表面改性方法。它是把工作(金属,合金,陶瓷等)放在离子注入机的真空靶室中,通过加高电压,把所需元素的离子注入到工件表层的一种工艺。材料经离子注入后,在其零点几微米的表层中增加注入元素和辐照损伤,从而使材料的物理化学性能发生显著变化。 大量实验证实,离子注入能使金属和合金的摩擦因素,耐磨性,抗氧化性,抗腐蚀性,耐疲劳性以及某些材料的超导性能,催化性能,光学性能等发生显著变化,能够大大提高材料的性能和使用寿命。离子注入在工业中应用能取得很好的效益,除延长工件的使用寿命外,还由于离子注入仅用较少量的合金元素,就可以得到较高的表面合金浓度,因而可以节约贵重金属[1]。 2.离子注入特点 与通常的冶金方法不同,离子注入是用高能量的离子注入来获得表面合金层的,因而有其特点: (1)离子注入是一个非热平衡过程,注入离子的能量很高,可以高出热平衡能量的2-3个数量级。因此,原则上周期表中的任何元素都可以注入任何基体材料。 (2)注入元素的种类,能量,剂量均可选择,用这种方法形成的表面合金,不受扩散和溶解度的经典热力学参数的限制,即可得到用其他方法难以获得的新合金相。 (3)离子注入层相对基体材料没有明显的界面,因此表面不存在粘附破裂或
铝金属标准曲线 铝金属标准曲线 序号X轴系数Y轴系数Y*轴系数 m/ug A A* 1 0.000 0.000 0 2 0.200 0.02 3 0.023 3 0.500 0.031 0.031 4 1.000 0.07 5 0.075 5 2.000 0.159 0.159 6 3.000 0.230 0.230 7 4.000 0.317 0.317 8 5.000 0.381 0.381 31 铝 31.1 铬天青S分光光度法 31.1.1 测定范围本法的最低检测限为0.125μg,若取25mL水样,则最低检测浓度为0.005mg/L,线性范围为0.005~0.200mg/L。一般水样中常见元素基本不干扰测定,铁、铜、锰含量高时,对测定有干扰,加入100μg/L抗坏血酸可消除25.0μg Cu,30.0μg Mn的干扰,10g/L巯基乙醇酸可消除25μgFe的干扰。31.1.2 方法提要在pH6.7~7.0范围内,铝在聚乙二醇辛基苯基醚和溴化十六烷基吡啶的存在下与铬天青S反应生成蓝色的四元体系混合胶束。比色定量。31.1.3 试剂 31.1.3.1 铬天青S溶液(1.0g/L):称取0.1g铬天青S(C23H13Cl2Na3O9S),溶于100mL乙醇溶液(1+1)中,混匀。 31.1.3.2 聚乙二醇辛基苯基醚溶液[ρ(聚乙二醇辛基苯基醚)=30%]:吸取乳化剂聚乙二醇辛基苯基醚(C34H62O11)3.0mL,溶于100mL水中。 31.1.3.3 溴化十六烷基吡啶溶液(3.0g/L):取0.6g溴化十六烷基吡啶 (C21H38BrN),溶于30mL乙醇中,加水稀释至200mL。 31.1.3.4 乙二胺 - 盐酸缓冲液(pH6.7~7.0):取无水乙二胺100mL,加水 200mL稀释,冷却后缓缓加入浓盐酸190mL,搅匀,用酸度计调节pH值为6.7~7.0,若pH大于7慢慢滴加浓盐酸,若pH小于6.7可补加乙二胺(1+2)溶液。 31.1.3.5 对硝基酚乙醇溶液(1.0g/L)。 31.1.3.6 氨水(ρ20=0.88g/mL)(1+6)。 31.1.3.7 硝酸[c(HNO3)=0.5mol/L]。 31.1.3.8 铝标准贮备溶液(1.0mg/mL):称取硫酸铝钾8.792g,溶于纯水中,定容至500mL,此溶液1.00mL含1.00mg铝。 31.1.3.9 铝标准使用溶液(1.0μg/mL):取1mL铝标准贮备溶液(31.1.3.8)进行定容至1000mL,此溶液1.00mL含1.00μg铝。 31.1.4 仪器 31.1.4.1 50mL具塞比色管(1+10硝酸浸泡)。 31.1.4.2 pHS-29A酸度计。 31.1.4.3 721分光光度计。 31.1.5 分析步骤
重金属检测方法汇总 重金属检测方法及应用 一、重金属的危害特性 从环境污染方面所说的重金属,实际上主要是指汞、镉、铅、铬、砷等金属或类金属,也指具有一定毒性的一般重金属,如铜、锌、镍、钴、锡等。我们从自然性、毒性、活性和持久性、生物可分解性、生物累积性,对生物体作用的加和性等几个方面对重金属的危害稍作论述。 (一)自然性: 长期生活在自然环境中的人类,对于自然物质有较强的适应能力。有人分析了人体中60多种常见元素的分布规律,发现其中绝大多数元素在人体血液中的百分含量与它们在地壳中的百分含量极为相似。但是,人类对人工合成的化学物质,其耐受力则要小得多。所以区别污染物的自然或人工属性,有助于估计它们对人类的危害程度。铅、镉、汞、砷等重金属,是由于工业活动的发展,引起在人类周围环境中的富集,通过大气、水、食品等进入人体,在人体某些器官内积累,造成慢性中毒,危害人体健康。 (二)毒性: 决定污染物毒性强弱的主要因素是其物质性质、含量和存在形态。例如铬有二价、三价和六价三种形式,其中六价铬的毒性很强,而三价铬是人体新陈代谢的重要元素之一。在天然水体中一般重金属产生毒性的范围大约在1~10mg/L之间,而汞,镉等产生毒性的范围在0.01~0.001mg/L之间。 (三)时空分布性: 污染物进入环境后,随着水和空气的流动,被稀释扩散,可能造成点源到面源更大范围的污染,而且在不同空间的位置上,污染物的浓度和强度分布随着时间的变化而不同。(四)活性和持久性: 活性和持久性表明污染物在环境中的稳定程度。活性高的污染物质,在环境中或在处理过程中易发生化学反应,毒性降低,但也可能生成比原来毒性更强的污染物,构成二次污染。如汞可转化成甲基汞,毒性很强。与活性相反,持久性则表示有些污染物质能长期地保持其危害性,如重金属铅、镉等都具有毒性且在自然界难以降解,并可产生生物蓄积,长期威胁人类的健康和生存。 (五)生物可分解性: 有些污染物能被生物所吸收、利用并分解,最后生成无害的稳定物质。大多数有机物都有被生物分解的可能性,而大多数重金属都不易被生物分解,因此重金属污染一但发生,治理更难,危害更大。 (六)生物累积性: 生物累积性包括两个方面:一是污染物在环境中通过食物链和化学物理作用而累积。二是污染物在人体某些器官组织中由于长期摄入的累积。如镉可在人体的肝、肾等器官组织中蓄积,造成各器官组织的损伤。又如1953年至1961年,发生在日本的水俣病事件,无机汞在海水中转化成甲基汞,被鱼类、贝类摄入累积,经过食物链的生物放大作用,当地居民食用后中毒。 (七)对生物体作用的加和性: 多种污染物质同时存在,对生物体相互作用。污染物对生物体的作用加和性有两类:一类是协同作用,混合污染物使其对环境的危害比污染物质的简单相加更为严重;另一类是拮抗作用,污染物共存时使危害互相削弱。 二、重金属的定量检测技术
我国环境中有机污染物分析方法及痕量富集技术的进展 王 梅,张莘民 (泰州市环境监测中心站,江苏 泰州 225300) 摘 要:综述了我国环境中有机污染物的分析方法和痕量富集技术,介绍了吹扫-捕集法、固相微萃取技术、固相萃取技术、半渗透膜采样技术与分析仪器联用在有机污染物测定中的应用情况。 关键词:有机污染物;分析方法;痕量富集技术;环境;中国 中图分类号:O656 文献标识码:A 文章编号:1006-2009(2004)01-0013-04 Development of Environmental Organic Pollutants .Analysis Method and Trace Enrichment Technology in China WANG Mei,ZHANG Xin -min (Taizhou Environmental Monitoring Center ,Taizhou ,Jiangsu 225300,China) Abstract:Development of environmental organic pollutants .analysis method and trace enrichment technology in China were discussed,such as blowing and tracing,solid phase micro -extracting technology,solid phase extracting technology,sem-i permeable membrane sampling technology. Key words:Organic pollutant;Analysis method;Trace enrichment technology;Environment;China 收稿日期:2003-07-07;修订日期:2003-11-11 作者简介:王 梅(1966)),女,江苏泰州人,工程师,大学,从事环境监测工作。 有机污染物对人类健康和生态平衡构成了严重威胁。经过20多年的努力,我国环境中有机污染物的分析方法和痕量富集技术取得了明显的进展,逐步接近了世界先进水平。1 有机污染物的分析方法 近年来,气相色谱(GC )、高效液相色谱(HPLC)、质谱、色质联用等技术在环境监测领域获得了广泛的应用,并取得了许多重要成果 [1-4] ,为 检测环境中的有机污染物开辟了广阔的前景。 我国从20世纪80年代开始采用GC 法作为大气中烃类和三氯乙醛、废气中苯系物的分析方法,1990年又将其定为空气和废气中20种有机化合物的分析方法,并用HPLC 法测定空气中的苯并(a)芘。在空气中有机污染物的监测分析方法中,色谱法占77.3%。 从1983年开始,我国先后将GC 法列为水中六六六、DDT 和苯系物的分析方法和标准方法,1989年规定以GC 法作为水中苯系物、氯苯类、六六六、DDT 、有机磷农药(总量)、三氯乙醛和硝基苯类等 有机物的分析方法[5],并首次采用HPLC 法分析水中16种多环芳烃。5水和废水监测分析方法6(第 4版)中,将GC 法作为苯系物、挥发性卤代烃、五氯酚、氯苯类、硝基苯类、有机氯农药、有机磷农药、阿特拉津、丙烯腈、三氯乙醛等有机物的分析方法;将GC/MS 法作为苯系物、挥发性卤代烃、五氯酚、二氯酚、邻苯二甲酸酯、己二酸酯、有机氯农药、多环芳烃、二恶英类、多氯联苯、有机锡等有机物的分析方法;将HPLC 法作为酚类、苯胺类、邻苯二甲酸酯类、阿特拉津、6种特定多环芳烃等有机物的分析方法。在水中有机污染物的监测分析方法中,色谱法占69.2%。2 痕量富集技术2.1 吹扫-捕集法 吹扫-捕集法是20世纪70年代中期推出的痕量挥发性有机化合物的富集方法,它具有灵敏度 ) 13)第16卷 第1期环境监测管理与技术2004年2月
版本1: 土壤中铜锌镉铬镍铅六中重金属全量一次消解测定方法.用氢氟酸-高氯酸-硝酸消解法,国家标准物质检测值和标准值吻合性很好,方便可行.具体方法: 准确称取0.5克土壤样品(过0.15mm筛)于四氟坩埚中,加7毫升硝酸+3毫升高氯酸+10毫升氢氟酸加盖,放置过夜(不过夜效果同),电热板上高温档加热(数显的控制温度300~350度)1小时,去盖,加热到近干,冷却到常温,然后再加3毫升硝酸+2毫升高氯酸+5毫升氢氟酸,高温档继续加热到完全排除各种酸,既高氯酸白烟冒尽,加1毫升(1+1)盐酸溶解残渣,完全转移到25毫升容量瓶中,加0.5毫升的100g/L的氯化铵溶液,定容,然后原子吸收分光光度计检测,含量低用石墨炉,注意定容完尽快检测锌,且锌估计需要适当的稀释.其实放置几天没有问题,相对比较稳定拉. 版本2: 1)称量0.5000g样品放入PTFE(聚四氟乙烯)烧杯中(先称量样品,后称量标 样),用少量去离子水润湿; 2)缓缓加入10.0mLHF和4.0mLHClO4(如果在开始加热蒸发前先把样品在混合 酸中静置几个小时,酸溶效果会更好一些),加盖后在电热板上200℃下蒸发(蒸发至样品近消化完后打开坩埚盖)至形成粘稠状结晶为止(2~3小时); 3)视情况而定,若有未消化完的样品则需要重新加入HF和HClO4,每次加入都 需要蒸发至尽干;若消化完全则直接进行下一步; 4)加入4.0mLHClO4,蒸发至近干,以除尽残留的HF; 5)加入10.0mL的5mol/L HNO3,微热至溶液清亮为止。检查溶液中有无被分解 的物料。如有,蒸发至近干,执行步骤4(此时可以酌情减半加酸); 6)待清亮的溶液冷却后,转入容量瓶,用去离子水定容至50mL(此时所得溶 液中硝酸含量为1mol/L),然后立即转移到新聚丙烯瓶中储存。 附: 现在一般做法是,砷汞用1+1的王水在沸水煮2小时,加固定剂(含5g/l重铬酸钾的5%硝酸溶液),在50毫升比色管中,固定,然后用原子荧光光谱仪测定砷汞.
游离氧化铁,游离氧化铝的测定 试剂: 1.连二亚硫酸钠 2.柠檬酸溶液(0.3mol/L)称取5个结晶水的柠檬酸钠104.4克溶于水,稀释至1L. 3.重碳酸钠溶液(NaHCO3=1mol/L) 称取84克碳酸氢钠溶于蒸馏水中,稀释至1L. 4.氯化钠溶液(1mol/L)称取氯化钠58.45克溶于蒸馏水,稀释至1L. 5.盐酸羟胺溶液(100克/L)称10克盐酸羟胺溶于蒸馏水,定容至100ML 6.邻啡罗啉显色剂(1克/L)称0.1克邻啡罗啉溶于100ML蒸馏水中,不溶可少许加热。 7.乙酸钠溶液(100克/L)称10克乙酸钠溶于蒸馏水中,定容至100ML 8.铁标准溶液:取纯金属铁粉0.1000克溶于稀盐酸中,加热溶之,冷却,洗入1000ML容量瓶中,定容 后摇匀,即为铁标准液(P(Fe=100mg/L)) 操作步骤: 1.游离氧化铁、铝的分离 称取过0.25MM筛(60目)土壤样品1.0-2.0克,置于50ML离心管中。加20ML柠檬酸溶液和2.5ML 重碳酸钠溶液,在水浴中加热至80℃,用小勺加入连二亚硫酸钠0.5克(估计量),不断搅动,维持15分钟。冷却后离心机分离,如分离不清,可加饱和氯化钠溶液5ML。将清液倾入50ML容量瓶中,如此重复处理1次至2次,此时离心管中的残渣是浅灰色或灰白色。最后用氯化钠溶液洗涤离心管中的残渣2次至3次。洗液一并倾入同一容量瓶中,定容,供测铁铝之用。 2.试铁灵铁铝联合比色法测定提取液中铁、铝 铝-试铁灵络合物在波长370NM时出现吸收,铁-试铁灵络合物则在600NM和370NM时均出现吸收。因此,试铁灵比色法就能在一个显色液中同时测定铁和铝。 1)硝酸溶液(1MOL/L)吸取63ML硝酸稀释至1L. 2)乙酸钠溶液(100克/L)称10克乙酸钠溶于蒸馏水中,定容至100ML,以PH计指示用NaOH或冰乙酸调至PH5.5. 3)试铁灵溶液:0.2克试铁灵试剂溶于100ML蒸馏水中。 4)铁标液的配制见上面的方法(已有铁标液,不需要同学配制) 5)铝标准溶液:称取金属铝片0.5000克,加15ML 盐酸(1:1)溶解,稀释至1L,铝的浓度为500MG/L. 再稀释至5MG/L备用,比色时,铝的色阶可采用0,0. 1,0.2,0.3,0.4,0.6,0.8,1MG/L. 3.取待测液(即上面游离氧化铁,铝分离方法中得到的待测液)10ML于25ML容量瓶中,加1.0ML硝酸 溶液,再加PH5.5的乙酸钠溶液6ML,试铁灵溶液2ML,用玻璃棒沾少许液体于PH试纸上,如果此时液体的PH值约为5.0-5.5,即可定容,如果不在此范围要调PH值。每次添加溶液均需摇混均匀。24
水样中各种重金属的测定方法 1铜、锌、铅、镉的测定火焰原子吸收法(水和废水监测分析方法第四版增补版pp.325-326) 本法适用于测定地下水、地表水、和废水中的铅锌铜镉。 仪器:原子吸收分光光度计 试剂:硝酸,优级纯;高氯酸,优级纯;去离子水; 金属标准储备液:准确称取经稀酸清洗并干燥后的0.5000g光谱重金属,用50ml(1+1)硝酸溶解,必要时加热直至溶解完全。用水稀释至500.0ml,此溶液每毫升含1.00mg金属。 混合标准容液:用0.2%硝酸稀释金属标准储备液配制而成,使配成的混合标准溶液每毫升含镉、铜、铅和锌分别为10.0、50.0、100.0、和10.0μg。 步骤 (1)样品预处理 取100ml水样放入200ml烧杯中,加入硝酸5ml,在电热板上加热消解(不要沸腾)。蒸至10ml左右,加入5ml硝酸和高氯酸2ml,再次蒸至1ml左右。取下冷却,加水溶解残渣,用水定容至100ml。 取0.2%硝酸100ml,按上述相同的程序操作,以此为空白值。(2)样品测定 据表1所列参数选择分析线和调节火焰。仪器用0.2%硝酸调零。吸入空白样和试样,测量其吸光度。扣除空白样吸光度后,从校准曲线上查出试样中的金属浓度。如可能,也从仪器中直接读出试样中的
金属浓度。 表1 元素分析线波长(nm)火焰类型本法测定范围(mg/L)镉228.8 乙炔-空气,氧化型0.05~1 铜324.7 乙炔-空气,氧化型0.05~5 铅283.3 乙炔-空气,氧化型0.2~10 锌213.8 乙炔-空气,氧化型0.05~1 (3)标准曲线 吸取混合标准溶液0, 0.50,1.00, 3.00,5.00和10.00ml,分别放入六个100ml容量瓶中,用0.2%硝酸稀释定容。此混合标准系列各重金属的浓度见表2。接着按样品测定的步骤测量吸光度,用经空白校正的各标准的吸光度对相应的浓度作图,绘制标准曲线。 表2 混合标准使用溶液体积 (ml) 0 0.50 1.00 3.00 5.00 10.00 标准系列各重金属浓度(mg/L)镉0 0.05 0.10 0.30 0.50 1.00 铜0 0.25 0.50 1.50 2.50 5.00 铅0 0.50 1.00 3.00 5.00 10.00 锌0 0.05 0.10 0.30 0.50 1.00 注:定容体积100ml 计算 被测金属(mg/L)= v m 式中:m—从校准曲线上查出或仪器直接读出的被测金属量(μg);
176 HUANJINGYUFAZHAN ▲ 安静 (东北大学 冶金学院 资源与环境系,辽宁 沈阳 110819) 摘要:随着抗生素在医学临床及养殖业中的大量应用,环境介质中抗生素的残留已成为普遍关注的环境问题。水环境是抗生素重要的归宿 地之一,本文从样品预处理以及检测分析两个方面系统分析了水环境中抗生素残留检测方法的原理、特点以及应用情况,并展望了该领域 未来的研究重点及发展方向。 关键词:水环境;抗生素;样品预处理;检测方法 中图分类号:X830.2 文献标识码:A 文章编号:2095-672X(2019)10-0176-03 DOI:10.16647/https://www.doczj.com/doc/5b8926103.html,15-1369/X.2019.10.100 Methods on detection of antibiotics residues in aquatic environment An Jing (Department of Resources and Environment, School of Metallurgy, Northeastern University, Shenyang Liaoning 110819, China) Abstract: Antibiotics residues in environmental media has attracted wide attention with the extensive application of antibiotics in clinical medicine and breeding industry,?and?aquatic?environment?is?one?of?the?important?destinations?of?antibiotics.?This?paper?systematically?analyzed?the?methods?of?sample?pretreatment?and?detection?of?antibiotic?residues?in?aquatic?environment.?The?principle,?characteristics?and?application?of?every?methods?were?summarized?and?the?future?research?emphasis?and?development?direction?in?this?field?were?also?proposed. Key words:Aquatic environment;Antibiotics;Sample pretreatment;Detection methods 抗生素广泛应用于医学临床及动物、水产养殖等领域,药物经各种给药途径进入动物体内后,不仅造成肉、蛋和乳等动物性食品中的残留,也会造成环境介质中的残留。水体已成为环境中抗生素最重要的归宿地之一,且目前已经在地表水、污水、养殖场废水甚至是地下水中检测到抗生素的存在。因此,加强水体抗生素检测、正确评估抗生素对主要水体的污染,同时提出科学、合理的管理方法显得极为迫切。这不仅关系到人民群众的身体健康,也对维持水域环境及水域生态系统的平衡、稳定具有重要的现实意义。 1?样品预处理方法 抗生素在水环境中的残留浓度一般属于微量或痕量级别,因此,在检测分析前需要对环境样品进行预处理,以对抗生素进行提取和纯化。 1.1?固相萃取 固相萃取是利用被萃取物质在液固两相间的分配作用进行样品前处理的一种分离技术。固相萃取以固体填料条充裕塑料小柱中作为固定相,样品溶液中被测物或干扰物吸附到固定相中,使被测物与样品机体或干扰组分得以分离[1]。固相萃取技术克服了萃取过程中容易乳化等缺点,不需要大量互不相溶的溶剂,且可同时完成样品的富集与净化,大大提高了检测灵敏度,并具有快速、可自动化批量处理以及重现性好等优点[2],是水环境中抗生素残留检测的最为常用的预处理方法。目前,固相萃取与色谱-质谱联用技术应用广泛,刘玉春等[3]应用此技术组合,建立了水中痕量大环内酯类抗生素的分析方法,加标纯水和实际水样的回收率在71%~111%之间,相对标准偏差在3.7%~8.6%之间,其定量下限为5ng/L。 1.2?固相微萃取 固相微萃取是在固相萃取技术上发展起来的一种微萃取分离技术,是一种集进样、萃取、浓缩功能于一体的样品制备技术,其原理是基于萃取涂层与样品之间的吸附(吸着)-解吸平衡[4]。萃取效率的高低取决于萃取纤维涂层的性质,通常根据待测物质的性质、分析方法的灵敏度、选择性以及重现性来选择萃取纤维涂层[5]。固相微萃取技术几乎可以用于气体、液体、生物、固体等样品中各类挥发性或半挥发性物质的分析。与固相萃取技术相比,固相微萃取操作更简单,携带更方便, 操作费用也更加低廉,另外克服了固相萃取回收率低、吸附剂孔道易堵塞的缺点,因此,成为目前所采用的样品前处理技术中应用最为广泛的方法之一。庄园等[6]以土霉素为模板分子制备了分子印迹固相微萃取涂层,建立了选择性萃取-高效液相色谱法同时测定牛奶和水样中四环素、盐酸土霉素和金霉素三种四环素类抗生素的分析方法,水样中三种抗生素的检出限为5~10μg/L,加标水平为500μg/L时,回收率范围为97.8%~109.0%,相对标准偏差为3.7%~6.4%。 1.3?磁性固相萃取 磁性固相萃取也称为磁纳米-微萃取技术,是以磁性或可磁化的材料作为吸附剂基质的一种分散固相萃取技术[7]。随着磁性吸附材料性能的不断完善,磁性固相萃取也发展成为样品预处理的重要方法。磁性固相萃取的出现,减少了有机溶剂的使用量,改变了常规固相萃取必须将萃取材料填充成柱的模式,解决了样品体积很大时常规固相萃取耗时较长的问题,更加易于实现自动化,并且可以对样品中的痕量化合物进行高倍的富集。XIAO等[8]提出了一种以二硫化钼-氧化石墨烯为载体的磁性纳米粒子(Fe 3 O 4 /Go/MoS 2 )作为磁性固相萃取的吸附剂,对水中的左氧氟沙星、帕珠沙星、加替沙星等抗生素进行分析的方法,制备的磁 性Fe 3 O 4 /Go/MoS 2 纳米复合材料对氟喹诺酮类抗生素有良好的富集能力,检测限为0.25~0.50ng/mL,水样分析回收率在85.6%~106.1%之间。刘小燕等[9]采用一步法制备了离子液体磁性石墨烯( IL@MGO),建立了磁性固相萃取-超高效液相色谱质谱法测定环境水体中的磺胺类抗生素的方法,6种抗生素的检出限为 0.75~1.47 ng/L,加标回收率在 86.4%~103.4%之间。 1.4?液相微萃取 液-液萃取耗时长,所需高纯溶剂量大,导致成本提高以及对环境的污染,且部分样品会发生乳化现象而影响测定,目前应用逐渐减少[10]。液相微萃取是在液-液萃取基础上,通过减少溶剂用量实现液-液萃取的微型化。该技术集样品采集、萃取、富集等过程于一体,具有操作简便、富集倍数高、成本低、环境友好等优点,被广泛应用于食品、药品
金属铝的测定方法1: 试剂: 1.硫酸铁溶液(20%),称取Fe2(SO4)3.9H2O 200克溶解于700mlH2O和75mlH2SO4中,溶毕冷却,用水稀释至1000ml。 2.磷酸(浓) 3.二苯胺磺酸钠(0.5%) 4.重铬酸钾标准溶液(0.1N),精确称取4.9028克经二次结晶并于110-130℃,干燥过的基准重铬酸钾于100ml水中溶解后,移入1000ml 容量瓶中,用水稀释至刻度,摇匀备用。测定方法: 称取0.2000克试样于300ml三角瓶中,加入50ml硫酸铁溶液,塞上带有长60-70cm玻璃管(或封闭漏斗)的塞子,置于沸水溶或低温电炉上微沸,加热至试样中金属铝全部溶解,取下,换上无孔橡皮塞塞紧,以冷水冷却至室温,加入10mlH3PO4,3滴0.5%二苯胺磺酸钠批示剂,用0.1N重铬酸钾标准溶液滴定至紫色为终点,记下消耗的重铬酸钾体积。 计算: Al%=N x V x26.98x100/3000C 其中:N-重铬酸钾的当量浓度 V-滴定所消耗的重铬酸钾体积(ml) 26.98-铝原子量(g) G-试样重量(g) 金属铝的测定方法2: 1.称取试样0.5000g于300ml锥型瓶中,加入FeCl3溶液(80g/L)50ml,放入磁棒,塞紧胶塞,置于恒控磁力搅拌器上搅拌40-60min,然后取下定容250ml,摇匀,用慢速滤纸干过滤。 2.分取滤液100ml于400ml烧杯中,加HCl(1+1)10ml,HClO420ml冒烟至近干,取下稍冷,加HCl(1+1)5ml,加水至约200ml,煮沸取下,用40%的NaOH调至PH=3-4,加入六次甲基四胺(30%)20ml,趁热过滤,将沉淀用热水和HCl(1+1)洗入400ml 烧杯中,然后用40%NaOH调至Fe(OH)3沉淀出现,再加入8gNaOH固体,煮沸1-2min,取冷却至室温,定容250ml,摇匀,干过滤。 3.分取滤液100ml于400ml烧杯中,加入过量EDTA溶液,酚酞2滴,用HCl(1+1)调至红色刚褪过去并过量3滴,加醋酸-醋酸钠缓冲溶液20ml,于电炉上煮沸3-5min,以PAN做指示剂,用CuSO4标准溶液滴至紫红色为终点,不记读数,加入0.5-1.0g NH4F,继续煮沸1-2min,取下,用CuSO4标准溶液滴至紫红色为终点,记下读数V1,并计算出结果,得到MAL的浸取率。 计算公式:MAL=V1x C x0.0270/G1x100 式中C-CuSO4标准溶液摩尔浓度,单位为摩尔每升(mol/L) V1-测定MAL时所消耗的标液体积,单位为毫升 G1-测定MAL时分取试液相当的试样重,单位为克
有机磷的测定方法 1.3.1气相色谱法 气相色谱法是一种以气体为流动相的柱色谱分离分析方法,它又可分为气液色谱法和气固色谱法。它的原理简单,操作方便。在全部色谱分析的对象中,约20%的物质可用气相色谱法分析。气相色谱法具有分离效率高、灵敏度高、分析速度快及应用范围广等特点。气相色谱法能分离性质极相似的物质,如同位素、同分异构体、对映体以及组成极复杂的混合物,如石油、污染水样及天然精油等。它的分离能力主要是通过选择高选择性固定相和增加理论塔板数来达到。气相色谱法使用高灵敏度的检测器,有的检测器其检测下限可达10-12—10-14g,是痕量分析不可缺少的工具之一。例如,它可检测食品中10-9数量级的农药残留量、大气污染中10-12数量级的污染物等。气相色谱法测定一个样品只需几分钟到几十分钟,分析速度很快,如用微机控制整个操作过程和数据处理系数,分析周期更短。在仪器允许的气化条件下,凡是能够气化且热稳定、不具腐蚀性的液体或气体,都可用气相色谱法分析。有的化合物因沸点过高难以气化或热不稳定而分解,则可以通过化学衍生化的方法,使其转变成易气化或热稳定的物质后再进样分析[2]。 只要在气相色谱仪允许的条件下可以气化而不分解的物质,都可以用气相色谱法分析。对部分热不稳定物质,或者难以气化的物质,通过化学衍生化的方法,仍可以用气相色谱法分析。所谓化学衍生化,就是通过合适的化学反应,使原先热不稳定性物质或难以气化物质转变成三甲基硅烷基衍生物或酯类、醚类等衍生物,以降低沸点和极性,增加稳定性和挥发度。 对被分析对象的分离,主要是选择良好的固定液及优化的操作条
件,而对固定液,不是只有唯一的选择[3]。下面举例说明用气相色谱法对不同类物质的分离。对低沸点烃类分离可以用角鲨烷柱或GDX类吸附剂;对高沸点烃类,可以用SE-30或OV-101类。分离醇、醚、醛、酯、酮、酸类等可以用PEG-20M。分离生物碱可以用SE-30,或OV类。分离氨基酸衍生物可以用OV类或XE-60。分离碳水化合物或糖类可以预先衍生化为三甲基硅烷基衍生物,用OV类、SE-30或XE-60。分离药物可以用OV类或DEGS。杀虫剂可以用SE-30、OV类或PEG-20M。分离萜类可以用SE-30、PEG-20M、OV类或DEGS。 分离比较简单的样品,可以用填充柱,分离比较复杂的样品应用开管柱,并采用程序升温方式。 1.3.2液相色谱法 21世纪初,液相色谱开始出现,但它的发展速度非常缓慢,在相当长的一段时间内,没有得到广泛应用。直到后来,出现了高效液相色谱后,液相色谱才得到了飞速发展,在各领域中广泛使用。它的复兴既借鉴了气相色谱的成功经验,也因气相色谱的缺点与不足而促进。液相色谱和气相色谱相比较,在以下几个方面具有优越性:气相色谱不适用于不挥发物质和对热不稳定物质,而液相色谱却不受样品的挥发性和热稳定性的限制。有些样品因为难以汽化而不能通过柱子,热不稳定的物质受热会发生分解,也不适用于气相色谱法。这使气相色谱法的使用范围受到了限制。据统计,目前气相色谱法所能分析的有机物,只占全部有机物的很少部分。另一方面,液相色谱却不受样品的挥发性和热稳定性的限制。所以液相色谱非常适合于分离生物、医药有关的大分子和离子型化合物,不稳定的天然产物,种类繁多的其它高分子及不稳定的化合物。对于很难分离的样品,用液相色谱常比用气相色谱容易完成分离,主要有以下三个方面的原因:液相色谱中,
生活饮用水标准检验方法金属指标铝铬天青S分光光度法 1.范围 本标准规定了用铬天青S分光光度法测定生活饮用水及其水源水中的铝。 本法适用于生活饮用水及其水源水中铝的测定。 本法的最低检测质量为0.20μg,若取25mL水样,则最低检测质量浓度为 0.008mg/L。 水中铜、锰、及铁干扰测定。1mL抗坏血酸(100g/L)可消除25μg铜、20μg锰的干扰。2mL巯基乙酸醇(10g/L)可消除25μg铁的干扰。 2.原理 在pH6.7~7.0范围内,铝在聚乙二醇辛基苯醚(OP)和溴代十六烷基吡啶(CPB)的存在下与铬天青S反应生成蓝绿色的四元胶束,比色定量。 3.试剂 3.1铬天青S溶液(1g/L) 3.2乳化剂OP溶液(3+100) 3.3溴代十六烷基吡啶(3g/L) 3.4乙二胺-盐酸缓冲液(pH6.7~7.0) 3.5氨水(1+6) )=0.5mol/L] 3.6硝酸溶液[c(HNO 3 3.7铝标准储备溶液[ρ(Al)=1mg/mL] 3.8铝标准使用溶液[ρ(Al)=1μg/mL] 3.9对硝基酚乙醇溶液(1.0g/L) 4.仪器 4.1具塞比色管:50mL,使用前需经硝酸(1+9)浸泡除铝。 4.2酸度计 4.3分光光度计 5.分析步骤 5.1取水样25.0mL于50mL具塞比色管中。
5.2另取50mL 比色管8支,分别加入铝标准使用溶液 0mL,0.20mL,0.50mL,1.00mL,2.00mL,3.00mL,4.00mL 和5.00mL ,加纯水至25mL 。 5.3向各管滴加1滴对硝基酚溶液,混匀,滴加氨水至浅黄色,加硝酸溶液至黄色消失,再多加2滴。 5.4加3.0mL 铬天青S 溶液,混匀后加1.0mL 乳化剂OP 溶液,2.0mLCPB 溶液,3.0mL 缓冲液,加纯水稀释至50mL ,混匀,放置30min 。 5.5于620nm 波长处,用2cm 比色皿以试剂空白为参比,测量吸光度。 5.6绘制标准曲线,从曲线上查出水样管中铝的质量。 6. 水样中铝的质量浓度计算 V m Al )(ρ 式中: )(ρAl ——水样中铝的质量浓度,单位为毫克每升(mg/L ) m ——从标准曲线上查的水样管中铝的质量,单位为微克(μg ) V ——水样体积,单位为毫升(mL ) 7. 精密度和准确度 5个实验室对浓度为20μg/L 和160μg/L 的水样进行测定,相对标准偏差均小于5%,回收率为94%~106%。
食品中几种常见的重金属检测方法 随着现阶段社会经济的快速发展,人们物质生活水平在不断提升,社会各界开始逐步重视食品安全问题。当前环境污染问题较为严重,各类重金属对食品安全构成了极大的威胁。为了有效应对食品安全中的重金属污染问题,当前需要对各类检测技术进行探究,促进食品安全检测工作质量的提升。 食品安全对于社会群众生命健康具有重要影响,当前相关食品检测机构需要从日常工作中提高责任意识,完善各项检测技术,确保食品安全。目前自然界中比重大于5的金属都被称为重金属,并不是所有的重金属都会对人体健康构成威胁,当重金属实际含量超出人体承受限度时会造成不同程度的危害,比如Pb、Cd、As、Hg等元素。许多重金属不能通过简单方法就能有效消除,如果人类长期使用被重金属污染后的食物,将会导致中毒问题。所以对重金属检测方法进行研究,对维护食品安全具有重要意义。 食物中常见重金属的主要来源概述 目前食品中存有的重金属来源主要有自然原因,也有诸多人为因素。自然原因主要包括不同地质和地理要素的影响,比如火山运动频繁的地区或是矿区,部分有毒重金属物质会对当地动植物产生不同程度污染,人类生活在此区域内,误食动植物都会诱发重金属中毒。人为因素导致的污染
主要是各类社会活动产生的主要后果,现阶段我国工业经济发展较快,各类工业生产活动会产生大量废渣和废水,此类废弃物当中存有较多重金属元素,如果相关部门不能对其进行有效处理,此类废弃物排放到自然环境中,不仅会破坏自然生态环境,还会对当地群众生命健康构成威胁。还有部分食物在实际存储和运输过程中与各类重金属元素进行直接接触,或是食物添加剂当中的有毒元素不断累积、发生相应化学反应都会导致重金属中毒现象的发生。 现阶段食品中几种常见的重金属检测方法探析 原子吸收光谱法。原子吸收光谱法主要是根据自由基础形态下的原子对辐射光进行共振吸收,通过光照强度来对食物中含有的重金属元素进行检测。此类方法实际操作较为便捷,能够最快速度得出相应结果,是当前食物重金属检测的重要技术。此类技术将磷酸二氢钾或是硝酸钯作为改进剂,通过添加改进剂能够使得原子温度有效降低,排除外界干扰因素,使得检测结果更加准确。现阶段在原子吸收光谱法中应用的吸收分光光度计都是通过微机进行控制,运用软件进行自动处理,简化了各项操作程序,有效缩短了实际反应时间。 原子荧光光谱法。原子荧光光谱技术是存在于原子发射和原子吸收之间的分析技术,在食物样品中添加还原剂,使得原子能够吸收特定的频率辐射,逐步形成激发态原子,此
《锆及锆合金化学分析方法第27部分:痕量杂质元素的测定-电感耦合等离子体质谱法》 编制说明 一、工作简况 1.1 任务来源及计划要求 根据国标委《国家标准委关于下达2017年第四批国家标准制修订计划的通知》(国标委综合〔2017〕128号)精神的文件精神,根据全国有色金属标准化技术委员会2018年3月14-3月16日昆明会议的“《锆及锆合金化学分析方法》标准任务落实会会议纪要”要求,批准由西北有色金属研究院负责起草国家标准《锆及锆合金化学分析方法第27部分:痕量杂质元素的测定电感耦合等离子体质谱法》国家标准,项目计划编号为20173516-T-610,项目要求2018年度完成。 1.2 调研和分析工作的情况 锆是一种重要的稀有金属,具有较高的抗腐蚀性能、极高的熔点、超高的硬度和强度等特性,被广泛用在航空航天、军工、核反应、原子能等领域。锆的热中子俘获截面小,有突出的核性能,是发展原子能工业不可缺少的材料,国内的大型核电站普遍都用锆材。用锆和锆合金作核潜艇的核燃料包套和压力管。 铝、硼、钴、铜、铬、铪、镁、锰、钼、镍、铅、锡、钛、铀、钒和钨作为锆及锆合金中的痕量杂质元素,目前尚无相应的国家和行业标准分析方法,随着我国核工业的快速发展,锆材及其加工产品日益增多,制定包括痕量杂质元素在内的《锆及锆合金化学分析方法》国家标准显得尤为迫切。该项目的完成对保证产品质量,完善我国核用锆材的研制、生产产业链和提高民用锆材的生产能力有积极的指导意义。 本研究经硝酸和氢氟酸低温加热溶解试料。用电感耦合等离子体质谱法测定铝、硼、钴、铜、铬、铪、镁、锰、钼、镍、铅、锡、钛、铀、钒和钨的含量;以内标法校正基体的影响。按工作曲线法计算各元素的质量浓度,以质量分数表示测定结果。其中铝、钴、铜、铬、铪、镁、锰、钼、镍、铅、锡、钛、铀、钒和钨的测定范围(质量分数)为0.0001%~0.010%;硼的测定范围(质量分数)为0.00005%~0.010%。方法灵敏度高、重现性好,实用性强。1.3 起草单位情况 西北有色金属金属研究院成立于1965年。是我国重要的稀有金属材料研究基地和行业技术开发中心、是国内稀有金属科研生产基地项目和稀有金属材料加工国家工程研究中心、金属多孔材料国家重点实验室、超导材料制备国家工程实验室、中国有色金属工业西北质量监督检验中心、层状金属复合材料国家地方联合工程研究中心等的依托单位,地处西安、宝鸡两地六区。研究院现有资产总值64.6亿元,仪器设备3000多台套,占地3428亩,正式职工2874人,其中科技人员近千余人,有中国工程院院士1人,教授、高工200多人,博士、硕士300余名。形成了以钛产业为主业,覆盖超导材料、金属纤维及制品、稀贵金属材料等产业的多元化格局,其产品广泛应用于航空、航天、航海、信息、电子、能源、环保等国民经济重要领域。 材料分析中心其前身可追溯至成立于1966年11月的西北有色金属研究院第三研究室(金属物理研究室)和第二研究室(化学分析研究室)。在四十多年的发展中,中心完成各类课题320项,获奖成果24项,其中省部级科技进步二等奖4项、三等奖9项,市局级科技进步一等奖1项、二等奖1项。制/修订国家、国家标准50多项;主持了《钛及钛合金化学分析方法》、《锆及锆合金化学分析方法》等标准方法,研制了《钛合金化学成分标准物质》一套,并获得科技部三等奖;申报专利10余项,发表论文500余篇。中心资质齐全,通过了CMA、CAL、CNAS、DiLAC认证,是全国(稀有金属)质量控制与评价实验室、中国有色金属工业西北质量监督检验中心、陕西省有色金属产品质量监督检验站、陕西省有色金属材料