
罗红霉素(Roxithromycin)是德国Roussel-Uclaf公司20世纪80年代初开发的十四元环口服用大环内酯类抗生素[1]。1987年在法国首次上市,国内1995年上市。其结构与红霉素类似,为红霉素的衍生物,具有口服吸收好、对胃酸稳定、生物利用度高、半衰期长、不良反应小等特点[2]。
有关罗红霉素血药浓度的测试方法,目前最常见的有微生物法、高效液相色谱(HPLC)法和高效液相色谱—串联质谱(HPLC-MS)法。微生物法测量血药浓度简单、快捷,测试成本相对较低,但测试结果受多种因素影响,变异大;HPLC法具有选择性高、精密度好的特点,但罗红霉素在紫外区吸收峰弱,该测定方法还不是最为优化的方法[3-4]。HPLC-MS法操作系统误差小,测定方法快速,灵敏度高,重现性好,红霉素和内标峰形良好,无杂峰干扰,优于HPLC法[5]。
1材料与方法
1.1药品与试剂受试制剂:罗红霉素胶囊(西南药业股份有限公司,批号:20090713,规格:每粒150mg);参比制剂:罗红霉素胶囊(浙江亚太药业股份有限公司,批号:081210,规格:每粒150 mg);罗红霉素标准品(中国药品生物制品检定所,批号:130557-200502,含量94.1%);克拉霉素内标物(中国药品生物制品检定所,批号:130558-200501,含量97.5%),乙腈、甲醇均为色谱纯(Burdick&Jackson公司),乙酸铵亦为色谱纯(天津市光复精细化工研究所),试验用水均为Millipore超纯水。
1.2仪器Waters-Acquity型超高效液相色谱质谱系统(配QBA235型TQ检测器,K07UPA468M型Sample Manager,K07UP8 450M型Binay solvent Manager及Masslynxv4.1色谱管理软件,Waters公司),色谱柱为Acquity UPLC BEH C18(1.7μm,
2.1mm×50mm)色谱柱,MC-210S型电子天平(精度0.01mg,Sartorius公司),Abbott Diagnostics Division离心机,XH-86多用途旋涡混合器(江苏阜宁县罗桥校办工厂),Millipore超纯水系统等。
1.3色谱质谱条件
1.3.1色谱条件采用Acquity UPLC BEH C18(1.7μm,
2.1mm×50mm)色谱柱,以乙腈-0.01%乙酸铵水溶液为流动相,梯度洗脱方式,乙腈比例在4min内从30%变为70%,流速为0.3mL/min,柱温为室温(35±1)℃,进样量为5μL。1.
3.2质谱条件罗红霉素质荷比(m/e)为837.53~158.15,克拉霉素质荷比(m/e)为748.48~590.30;毛细管电压:3.5kV;锥孔电压:罗红霉素40V,克拉霉素40V;碰撞电压:罗红霉素30V,克拉霉素40V;脱溶剂温度:350℃;离子源温度:120℃;脱溶剂气流:600L/h;锥孔气流:50L/h。
1.4血浆样品的处理精密移取0.2mL空白血浆于1.5mL离心管中,加入内标溶液(25.00μg/mL)50μL,混旋1min,加碳酸钠(0.05mol/mL)100μL混匀5min后,加乙酸乙酯500μL混匀10min,离心5min,取上清液,氮气吹干后,用500μL流动相溶解后再次离心5min,去上清液进样。
1.5测定方法确证
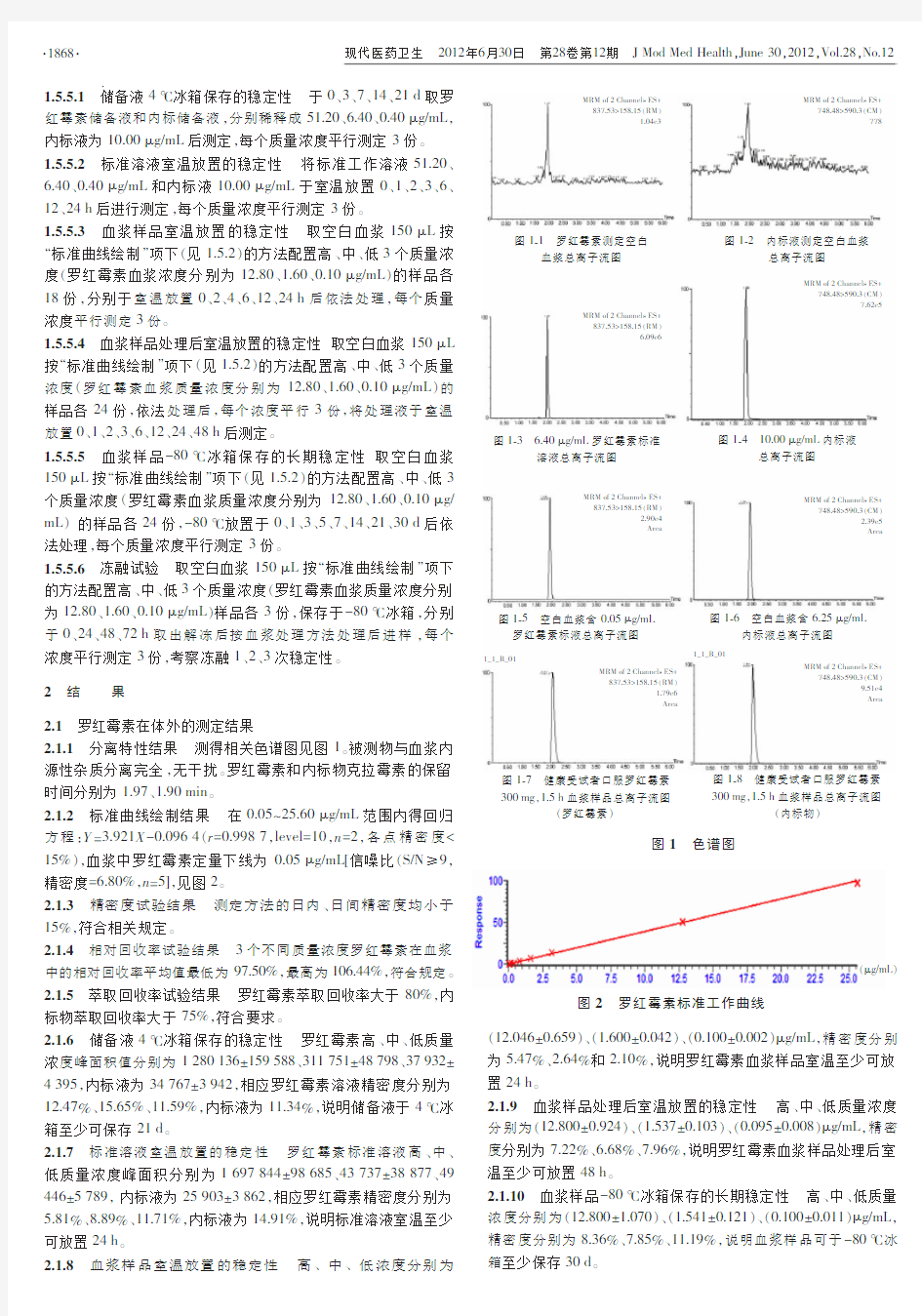
1.5.1分离特性在选定的色谱条件下,分别考察了7个不同来源的空白血浆、空白血浆加对照品、对照品及实测样品的色谱分离情况。
1.5.2标准曲线绘制取上述配置好的工作液50μL于0.15mL 血浆中,加入内标溶液(25.00μg/mL)50μL后按血浆处理方法配制成血浆质量浓度为25.60、1
2.80、6.40、
3.20、1.60、0.80、0.40、0.20、0.10、0.05μg/mL的溶液进样分析,每个质量浓度平行2份,进样量为3μL。记录质谱图,以质量浓度为横坐标,以罗红霉素与内标峰峰面积比为纵坐标进行线性回归。
1.5.3精密度试验取空白血浆150μL按“标准曲线绘制”项下(见1.5.2)的方法配置低、中、高3个质量浓度(罗红霉素血浆质量浓度分别为0.10、1.60、1
2.80μg/mL)的血浆样品各6份,按“血浆样品的处理”项下(见1.4)依法操作,于同1天不同时间段测定,计算日内精密度。每天低、中、高3个质量浓度的血浆样品各6份,连续测定3d,计算日间精密度。
1.5.4回收率试验
1.5.4.1相对回收率取空白血浆150μL按“标准曲线绘制”项下(见1.5.2)的方法配置低、中、高3个质量浓度(罗红霉素血浆质量浓度分别为0.10、1.60、1
2.80μg/mL)的样品各6份,并依法测定相对回收率。
1.5.4.2萃取回收率配制高、中、低3个质量浓度的标准工作液(5.12、0.64、0.04μg/mL),以及内标工作液
2.50μg/mL,每个质量浓度平行6份,以对应血浆药物质量浓度峰面积值除以标准溶液峰面积平均值计算萃取回收率。
1.5.5稳定性试验
罗红霉素胶囊人体生物等效性研究
汪潜1,李慧慧1,孙克宏1,张颖1,辛龙涛1,张娅1,郑飞鸣2(1.太极集团有限公司,重庆401147;
2.第三军医大学新桥医院,重庆400037)
【摘要】目的研究两种罗红霉素胶囊在健康受试者体内的药代动力学与相对生物利用度,评价其生物等效性。方法将健康受试者20例随机分成两组,每组10例,采用双交叉给药方案,分别口服受试胶囊和参比胶囊各300mg,于规定时间取血,以克拉霉素为内标,以超高效液相色谱-串联质谱法测定人血浆中罗红霉素浓度,所得数据经BECS软件处理,对血浆药物浓度-时间曲线下面积0-t(AUC0-t)、血浆药物浓度、时间曲线下面积0-∞(AUC0-∞)、达峰浓度(C max)进行方差分析和双单侧t检验,对达峰时间(T max)进行非参数检验。结果受试胶囊和参比胶囊T max分别为(2.367±1.295)、(2.134±1.537)h,C max分别为(7.702±1.902)、(7.920±1.957)μg/mL,AUC0-t分别为(103.77±31.46)、(103.10±31.76)μg/(h·mL),AUC0-∞分别为(110.05±35.25)、(107.97±34.72)μg/(h·mL),差异无统计学意义。结论经过对比研究2种罗红霉素胶囊人体内药代动力学和相对生物利用度,受试制剂和参比制剂生物等效。
【关键词】罗红霉素;药代动力学;生物利用度;高效液相色谱-质谱法
文章编号:1009-5519(2012)12-1867-03中图法分类号:R978.15文献标识码:B
通讯作者:辛龙涛(E-mail:xinlongtao@https://www.doczj.com/doc/3417872083.html,)。
亲爱的朋友,很高兴能在此相遇!欢迎您阅读文档罗红霉素软胶囊说明书,这篇文档是由我们精心收集整理的新文档。相信您通过阅读这篇文档,一定会有所收获。假若亲能将此文档收藏或者转发,将是我们莫大的荣幸,更是我们继续前行的动力。 罗红霉素软胶囊说明书 罗红霉素软胶囊(天迅)用于化脓性链球菌引起的咽炎及扁桃体炎,敏感菌所致的鼻窦炎、中耳炎、急性支气管炎,肺炎支原体或肺炎衣原体所致的肺炎;沙眼衣原体引起的尿道炎和宫颈炎;敏感细菌引起的皮肤软组织感染。下面是我们整理的,欢迎阅读。 罗红霉素软胶囊商品介绍 通用名:罗红霉素软胶囊 生产厂家:浙江维康药业有限公司 批准文号:国药准字Hxx0302 药品规格:0.15g*12粒 药品价格:¥24元 【通用名称】罗红霉素软胶囊 【商品名称】罗红霉素软胶囊(天迅) 【英文名称】RoxithromycinSoftCapsules
【拼音全码】LuoHongMeiSuRuanJiaoNang(TianXun) 【主要成份】罗红霉素软胶囊(天迅)主要成分为罗红霉素。 化学名:9—[O—[(2-甲氧基乙氧基)甲基]肟基]红霉素 分子式:C41H76N2O15 分子量:837.03 【性状】罗红霉素软胶囊(天迅)内容物为白色至淡黄色的混悬液。 【适应症/功能主治】化脓性链球菌引起的咽炎及扁桃体炎,敏感菌所致的鼻窦炎、中耳炎、急性支气管炎,肺炎支原体或肺炎衣原体所致的肺炎;沙眼衣原体引起的尿道炎和宫颈炎;敏感细菌引起的皮肤软组织感染。 【规格型号】0.15g*12s 【用法用量】空腹口服,一般疗程为5~12日。儿童可直接服用,或剪开囊壳将药液滴入饮料或牛奶中服用。成人:一次0.15g,一日2次;也可一次0.3g,一日1次。儿童:一次按体重2.5~5mg/kg,一日2次。 【不良反应】主要不良反应为腹痛、腹泻、恶心、呕吐等胃肠道反应,但发生率明显低于红霉素。偶见皮疹、皮肤瘙痒、头昏、头痛、肝功能异常(ALT及AST升高)、外周血细胞下降等。 【禁忌】对罗红霉素软胶囊(天迅)中任何成份、红霉素或其
总局关于发布人体生物等效性试验豁免指导原则的通告(2016 年第87号) 2016年05月19日发布为规范仿制药质量和疗效一致性评价工作,根据《国务院办公厅关于开展仿制药质量和疗效一致性评价的意见》(国办发〔2016〕8号)的有关要求,国家食品药品监督管理总局组织制定了《人体生物等效性试验豁免指导原则》,现予发布。 特此通告。 附件:人体生物等效性试验豁免指导原则 食品药品监管总局 2016年5月18日附件 人体生物等效性试验豁免指导原则 本指导原则适用于仿制药质量和疗效一致性评价中口服固体常释制剂申请生物等效性(Bioequivalence)豁免。该指导原则是基于国际公认的生物药剂学分类系统(Biopharmaceutics Classification System,以下简称BCS)起草。
一、药物BCS分类 BCS系统是按照药物的水溶性和肠道渗透性对其进行分类的一个科学架构。当涉及到口服固体常释制剂中活性药物成分(Active Pharmaceutical Ingredient,以下简称API)在体内吸收速度和程度时,BCS系统主要考虑以下三个关键因素,即:药物溶解性(Solubility)、肠道渗透性(Intestinal permeability)和制剂溶出度(Dissolution)。 (一)溶解性 溶解性分类根据申请生物等效豁免制剂的最高剂量而界定。当单次给药的最高剂量对应的API在体积为250ml(或更少)、pH值在1.0—6.8范围内的水溶性介质中完全溶解,则可认为该药物为高溶解性。250ml的量来源于标准的生物等效性研究中受试者用于服药的一杯水的量。 (二)渗透性 渗透性分类与API在人体内的吸收程度间接相关(指吸收剂量的分数,而不是全身的生物利用度),与API在人体肠道膜间质量转移速率直接相关,或者也可以考虑其他可以用来预测药物在体内吸收程度的非人体系统(如使用原位动物、体外上皮细胞培养等方法)对渗透性进行分类。当一个口服药物采用质量平衡测定的结果或是相对于静脉注射的参照剂量,显示在体内的吸收程度≥85%以上(并且有证据证明药物在胃肠道稳定性良好),则可说明该药物具有高渗透性。 (三)溶出度
附件1 生物等效性研究的统计学指导原则 一、概述 生物等效性(Bioequivalence, BE)研究是比较受试制剂(T)与参比制剂(R)的吸收速度和吸收程度差异是否在可接受范围内的研究,可用于化学药物仿制药的上市申请,也可用于已上市药物的变更(如新增规格、新增剂型、新的给药途径)申请。 目前生物等效性研究通常推荐使用平均生物等效性(Average Bioequivalence, ABE)方法。平均生物等效性方法只比较药代动力学参数的平均水平,未考虑个体内变异及个体与制剂的交互作用引起的变异。在某些情况下,可能需要考虑其他分析方法。例如气雾剂的体外BE研究可采用群体生物等效性(Population Bioequivalence,PBE)方法,以评价制剂间药代动力学参数的平均水平及个体内变异是否等效。 本指导原则旨在为以药代动力学参数为终点评价指标的生物等效性研究的研究设计、数据分析和结果报告提供技术指导,是对生物等效性研究数据资料进行统计分析的一般原则。在开展生物等效性研究时,除参考本指导原则的内容外,尚应综合参考《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》和《药物临床试验的生物统计学指导原则》等相关指导原则。 二、研究设计 (一)总体设计考虑 生物等效性研究可采用交叉设计或者平行组设计。 —1 —
1.交叉设计 生物等效性研究一般建议采用交叉设计的方法。交叉设计的优势包括:可以有效减少个体间变异给试验评价带来的偏倚;在样本量相等的情况下,使用交叉设计比平行组设计具有更高的检验效能。 两制剂、两周期、两序列交叉设计是一种常见的交叉设计,见表1。 表1 两制剂、两周期、两序列交叉设计 序列 周期 1 2 1 T R 2 R T 如果需要准确估计某一制剂的个体内变异,可采用重复交叉设计。重复交叉设计包括部分重复(如两制剂、三周期、三序列)或者完全重复(如两制剂、四周期、两序列),见表2和表3。 表2 两制剂、三周期、三序列重复交叉设计 序列 周期 1 2 3 1 T R R 2 R T R 3 R R T —2 —
罗红霉素使用说明书 【篇一:罗红霉素颗粒说明书】 罗红霉素胶囊说明书 【药品名称】 通用名:罗红霉素胶囊 英文名:roxithromycin dispersible capsules 汉语拼音:luohongmeisu jiaonang 本品主要成分为罗红霉素, 化学名为9—[o—[(2-甲氧基乙氧基)-甲基]肟]红霉素。 分子式:c41h76n2o15 分子量:837.03 【性状】本品为胶囊剂。 【药理毒理】本品为半合成的14元环大环内酯类抗生素。抗菌谱 与抗菌作用基本上与红霉素相仿,对革兰阳性菌的作用较红霉素略差,对嗜肺军团菌的作用较红霉素强。对肺炎衣原体、肺炎支原体、溶脲脲原体的抗微生物作用与红霉素相仿或略强。本品可透过细菌 细胞膜,在接近供体(“p”位)与细菌核糖体的50s亚基成可逆性结合,阻断了转移核糖核酸(t-rna)结合至“p”位上,同时也阻断了多 肽链自受位(“a”位)至“p”位的转移,因而细菌蛋白质合成受到抑制。 【药代动力学】口服吸收好,血药峰浓度(cmax)高,单剂量口服罗 红霉素150 mg后约2小时达血药峰浓度(cmax)6.6~7.9 mg/l,进食可使生物利用度下降约一半。分布广,扁桃体、鼻窦、中耳、肺、痰、前列腺及其他泌尿生殖道组织中的药物浓度均可达有效治疗水平。其蛋白结合率在血浓度2.5 mg/l时为96%。以原形及5个代谢 物从体内排出,7.4%自尿液排出。血消除半衰期(t1/2?)为8.4~15.5小时。 【适应症】本品适用于化脓性链球菌引起的咽炎及扁桃体炎,敏感 菌所致的鼻窦炎、中耳炎、急性支气管炎、慢性支气管炎急性发作,肺炎支原体或肺炎衣原体所致的肺炎;沙眼衣原体引起的尿道炎和 宫颈炎;敏感细菌引起的皮肤软组织感染。 【用法用量】 空腹口服,一般疗程为5~12日。 成人一次150mg,一日2次;也可一次300mg,一日1次。
为了避免大家在生物等效性试验备案的过程中少走弯路,帮助大家更顺利的通过,今天就为大家详细的讲解一下生物等效性试验备案的流程吧: (一)注册申请人向具有资质的药物临床试验机构提出申请,获得该机构伦理委员会的批准,并签署BE试验研究合同。 (二)注册申请人开展生物等效性试验前30天,应当在国家食品药品监督管理总局指定的化学药BE试验备案信息平台进行化学药BE试验备案,按要求提交备案资料。 提前30天申请,但未明确说30天未收到异议即可开展BE研究,这点很重大! (三)备案资料主要包括注册申请人信息、产品基本信息、处方工艺、质量研究和质量标准、参比制剂基本信息、稳定性研究、原料药、试验方案设计、伦理委员会批准证明文件等。 此条对讨论稿中的“合法原料”做了终版解释,无“合法原料”说法,所以可解读为新3+5,老3+6都是可以备案的啦。 (四)注册申请人BE试验的参比制剂及各参与方的基本信息等向社会公开。 (五)注册申请人在获得备案号后,应在第1例受试者入组前在国家食品药品监督管理总局药物临床试验登记与信息公示平台完成开展试验前的所有信息登记,并由国家食品药品监督管理总局向社会公示;1年内未提交受试者入组试验信息的,注册申请人须说明情况;2年内未提交受试者入组试验信息的,所获得备案号自行失效。 (六)注册申请人应严格执行《药物临床试验质量管理规范》(GCP),按照试验方案开展BE试验。BE试验过程中,参比制剂、原料药、制剂处方、工艺等发生变更,注册申请人应停止试验,通过备案平台提交试验中止的申请,国家食品药品监督管理总局将公示其中止试验。注册申请人根据变更情况,向国家食品药品监督管理总局提交备案变更资料,生成新的备案号后重新开展BE试验。 这条写的很好,BE试验过程中的终止与变更必须引起大家的关注,BE试验在国外的一次性通过几率有多少?偷偷做人体预试验这个事儿,靠谱不? (七)注册申请人应当在BE试验完成或因故终止一年内,在备案平台提交BE试验的总结报告或情况说明。 (八)注册申请人完成BE试验后,应将试验数据申报资料、备案信息及变更情况提交国家食品药品监督管理总局,在此基础上提出相应药品注册申请。注册申请人要承诺其注册申请资料及数据的真实、完整、规范。 BE结束后,才是正式的“药品注册申请”啦,在此之前都是企业自行验证药品质量的过程。而真实性的关注将化作永恒。 (九)未按本公告规定备案而开展的BE试验,国家食品药品监督管理总局不受理其注册申请。
生物等效性实验生物样品处理注意事项(严)
生物等效性实验生物样品处理注意事项一、样品采集后的的处理和贮存 鉴于生物样本的特点,为了避免样品中被测药物发生分解或产生其他化学变化,取样后最好立即进行分析测定,但实际工作中几乎无法做到,常需将收集到的样品冷藏、冰冻,临用前再融化并放至室温后使用。在样本冷冻贮藏前,需及时进行处理。 1.1血液样本处理注意事项 1.1.1. 在肌肉注射或静脉输含有葡萄糖或电解质(含钾、钠、氯离子)的液体时,建议3小时以后采集静脉血样本进行这些项目的检验,以防止上述检验项目因输液引起的假性升高。 1.1.2保定非麻醉状态的动物时应尽量避免用力挤压动物头颈和胸腹,以免引起血液淤滞,局部组织缺氧,造成血液某些成分的改变,特别是测定乳酸,血液含氧量等指标时。 1.1.3血液中红细胞内外成分有很大差异,溶血可造成红细胞内的物质向细胞外转移,如K+、Mg2+和某些酶类(LD、AST、ALT、ACP);另外,溶血还可干扰某些化学项目(TBil、DBil、TC等)的测定,严重影响结果的准确性,血样本应防止溶血。引起溶血的原因有:注射器采血时抽吸力太大;血液与抗凝剂比例失调;混匀样本时过度振荡;注射器或采血容器带水或容器污染;全血放置时间长或突然受冷或受热;注射器中的血沫注入采血容器;真空采血时如未
采满至相应刻度,残存负压造成红细胞破裂;不拔针头直接注入采血容器;样本离心时离心力过大等。为避免溶血,取血时应注意: ①、抽拉注射器时应尽量避免注射器内产生大量真空; ②、添加抗凝剂后的容器在除必要干燥流程后应及时密封; ③、混匀样本时避免用力过度,切勿产生泡沫; ④、避免重复使用注射器、针头、采血管、毛细玻璃管等一次性用品,手术刀片和剪刀等器材取材时尽量洗去残留血液; ⑤、采血时的室温应控制在22℃至25℃,采取的血液容器在需要放入冰盒时,切勿紧贴冰袋,冰水; ⑥、当注射器内因吸入空气产生血沫时,注意弃掉血沫,在将血液注入采血容器时勿将血沫一并注入; ⑦、使用真空采血管需抽取负压时切勿过量; ⑧、将血液注入采血容器时要除去针头,轻柔推入; ⑨、离心带有血细胞的血样时,按照规格设定离心参数; 1.1.4. 正确选择采集管。通常情况下多采用血清为样本(不抗凝),部分检测项目需注意样本属性为血清或血浆,两者不可替代。一些特殊检验项目需要使用抗凝剂时,应注意选择合适的抗凝剂并注意抗凝剂与血液的比例,以防止样本凝血或红细胞形态的改变;抗凝血样本采集后立即轻轻摇匀至少上下颠倒8次,以防凝血发生。 1.1.5. 多项化验采血顺序:血培养瓶(厌氧瓶优先)→蓝帽管→黑帽管→红/黄帽管→绿帽管→紫帽管→灰帽管→其他。
精心整理附件3 以药动学参数为终点评价指标的 化学药物仿制药人体生物等效性研究 技术指导原则 体循环的过程,通常将受试制剂在机体内的暴露情况与参比制剂进行比较。 在上述定义的基础上,以药动学参数为终点评价指标的生物等效性研究又可表述为:通过测定可获得的生物基质(如血液、血浆、血清)中的药物浓度,取得药代动力学参数作为终点指标,藉此反映药物释放并被吸
收进入循环系统的速度和程度。通常采用药代动力学终点指标C max和AUC 进行评价。 如果血液、血浆、血清等生物基质中的目标物质难以测定,也可通过测定尿液中的药物浓度进行生物等效性研究。 药效动力学研究: 2)两 每位受试者依照随机顺序接受受试制剂和参比制剂。对于半衰期较长的药物,可选择第2种试验设计,即每个制剂分别在具有相似人口学特征的两组受试者中进行试验。第3种试验设计(重复试验设计)是前两种的备选方案,是指将同一制剂重复给予同一受试者,可设计为部分重复(单制剂重复,即三周期)或完全重复(两制剂均重复,即四周期)。重复试验设
计适用于部分高变异药物(个体内变异≥30%),优势在于可以入选较少数量的受试者进行试验。 对于高变异药物,可根据参比制剂的个体内变异,将等效性评价标准作适当比例的调整,但调整应有充分的依据。 (二)受试者选择 18 60岁 通常推荐采用单次给药药代动力学研究方法评价生物等效性,因为单次给药在评价药物释放的速度和程度方面比多次给药稳态药代研究的方法更敏感,更易发现制剂释药行为的差异。 (五)稳态研究 若出于安全性考虑,需入选正在进行药物治疗,且治疗不可间断的患
者时,可在多次给药达稳态后进行生物等效性研究。 (六)餐后生物等效性研究 食物与药物同服,可能影响药物的生物利用度,因此通常需进行餐后生物等效性研究来评价进食对受试制剂和参比制剂生物利用度影响的差异。 2小 推荐采用实测药物峰浓度C max评价吸收速度。药物浓度达峰时间T max 也是评价吸收速度的重要参考信息。 2.吸收程度/总暴露量 对于单次给药研究,建议采用如下两个参数评价吸收程度: (1)从0时到最后一个浓度可准确测定的样品采集时间t的药物浓
罗红霉素胶囊治疗感染性疾病的效果分析 发表时间:2019-07-24T14:21:30.063Z 来源:《医药前沿》2019年17期作者:谭万江刘书红刘林高伟曹骊袁洪贺姝姝 [导读] 罗红霉素胶囊一般主要用于治疗皮肤、耳喉鼻、生殖器或呼吸道等部位因为革兰氏阳性菌或衣原体等敏感菌株感染所造成的感染问题。 (贵州省黔西南州中医院贵州黔西南 562400) 【摘要】选取2018年1月—12月来我院接受临床治疗的84例感染性疾病患者,随机分为观察组42例和对照组42例,对照组服用克拉霉素,观察组口服罗红霉素胶囊治疗。接受治疗一周时间后,观察两组治疗效果。结果:观察组治疗总有效率为95.24%高于对照组的 78.57%,组间对比,差异显著(P<0.05)。观察组不良反应发生率低于对照组,差异显著(P<0.05)。结论:应用罗红霉素胶囊,治疗感染性疾病可以获得良好的治疗效果,且不良反应较少。 【关键词】罗红霉素胶囊;临床应用;不良反应 【中图分类号】R453 【文献标识码】A 【文章编号】2095-1752(2019)17-0123-01 在临床上用药方面,罗红霉素胶囊一般主要用于治疗皮肤、耳喉鼻、生殖器或呼吸道等部位因为革兰氏阳性菌或衣原体等敏感菌株感染所造成的感染问题。但是随着罗红霉素胶囊这种抗生素在临床上的广泛应用,极大地增加了其出现不良反应的概率。本文选择2018年1月—12月间于我院接受临床治疗的84例感染性疾病患者为研究对象,对罗红霉素胶囊的临床应用效果及不良反应情况进行了对比分析,现报告如下。 1.资料与方法 1.1 一般资料 选择2018年1月—12月间于我院接受临床治疗的84例感染性疾病患者为研究对象。经临床确诊分析,全部患者均是由敏感菌所造成的生殖系统、皮肤、呼吸道或五官等感染患者。在感染性疾病类型方面,24例皮肤感染患者,16例泌尿生殖感染患者,28例五官感染患者,16例呼吸道感染患者。按照随机数字表法,随机分为观察组(42例)和对照组(42例)。在观察组中的42例患者中,男20例,女22例;年龄6~77岁,平均(42.14±1.35)岁。在对照组中的42例患者中,男19例,女23例;年龄5~77岁,平均(42.16±1.33)岁。两组患者在感染性疾病类型等一般资料方面差异不显著(P>0.05),具有可比性。 1.2 方法 对照组中的42例患者在临床治疗期间服用克拉霉素进行治疗,具体就是采用服用克拉霉素的方式进行治疗,成人按照0.5~1g/d,儿童按照7.5mg/kg,2次/d。观察组中的42例患者均行罗红霉素治疗,具体就是采取口服罗红霉素胶囊的方式来进行治疗,成人按照150mg/次,2次/2剂量服用;儿童按照2.5~5.0mg/kg每天,分2次进行药物服用,并分别在饭前1h或饭后4h进行服用。在两组患者在接受临床治疗一周后,对比分析两组患者的临床治疗情况。 1.3 评价指标 在本次研究中,罗红霉素胶囊临床应用疗效的观察指标主要包括患者感染性疾病的治疗情况以及患者在接受临床治疗期间出现的腹泻等不良反应情况。 1.4 统计学分析 采取SPSS20.0统计软件进行数据处理和分析,计量资料用(x-±s)表示,行t检验;计数资料用%表示,行χ2检验。P<0.05表示比较差异显著,具有可比性。 2.结果 2.1 两组患者临床治疗效果比较 在两组患者接受治疗一段时间后,观察组患者的临床治疗总有效率为95.24%,显著大于对照组的78.57%,组间比较差异显著(P<0.05) 2.2 两组患者不良反应情况比较 在两组患者接受治疗一段时间后,观察组患者的总不良反应率为4.76%,显著小于对照组的23.80%,组间比较差异显著(P< 0.05)。 3.讨论 3.1 罗红霉素胶囊及其应用机理 罗红霉素胶囊是一类新型的抗生素,属于大环内酯类范畴,是一种红霉素的衍生物。在应用罗红霉素胶囊期间,其可以有选择性地结合各类致病菌的70S核糖体的50S亚基,使其原有的构象发生改变,最终会对核糖体当中肽链转移酶的转位或活性等产生抑制作用,使诱发感染问题的致病菌的依赖性蛋白或RNA出现合成受阻问题,最终可以达到抗菌的效果。罗红霉素胶囊可以对大部位G+菌进行有效抑制,其对嗜血流干杆菌、肺炎球菌等的抗菌活性同克拉霉素之间大体相同,尤其是对衣原体、支原体以及军团菌等菌体的抗菌效果非常有效。罗红霉素胶囊具有吸收性好、穿透性强、耐酸性强等应用优势。 3.2 罗红霉素胶囊的常见应用及不良反应 目前,罗红霉素胶囊在临床上的应用主要是治疗那些因为支原体或衣原体等敏感菌所造成的肺炎、支气管炎、泌尿系统或皮肤感染等,相应地临床治疗效果也比较理想。然而,在临床应用罗红霉素胶囊期间却同样非常容易因为该种抗生素应用的增加而出现腹泻等不良反应,影响了患者的临床治疗效果。针对这种情况,在临床治疗患者过程中需要高度重视做好罗红霉素胶囊应用后的不良反应预防。比如,在临床治疗呼吸道感染患者期间,如果可以联合地运用糖皮质激素类治疗药物,那么可以有效地消除临床疾病的各种症状或体征,同时可以对患者的心肺功能进行改善,缓解其可能伴有的咳嗽等疾病病症;罗红霉素胶囊可以不受患者胃酸环境的影响,所以可以提升其消除幽门杆菌等感染菌的治疗效果。随着罗红霉素胶囊这种抗生素在临床上的广泛应用,极大地增加了其出现不良反应的概率,所以临床中要注意做好不良反应的防范工作。 在本次研究中,对比分析了克拉霉素和罗红霉素胶囊的临床应用效果及不良反应情况。在临床治疗总有效率和总不良反应率方面,观
附件3 以药动学参数为终点评价指标的 化学药物仿制药人体生物等效性研究 技术指导原则 一、概述 本指导原则主要阐述以药动学参数为终点评价指标的化学药物仿制药人体生物等效性试验的一般原则,适用于体内药物浓度能够准确测定并可用于生物等效性评价的口服及部分非口服给药制剂(如透皮吸收制剂、部分直肠给药和鼻腔给药的制剂等)。进行生物等效性试验时,除本指导原则外,尚应综合参考生物样品定量分析方法验证指导原则等相关指导原则开展试验。 生物等效性定义如下:在相似的试验条件下单次或多次给予相同剂量的试验药物后,受试制剂中药物的吸收速度和吸收程度与参比制剂的差异在可接受范围内。生物等效性研究方法按照研究方法评价效力,其优先顺序为药代动力学研究、药效动力学研究、临床研究和体外研究。 药代动力学(药动学)研究: 对于大多数药物而言,生物等效性研究着重考察药物自制剂释放进入体循环的过程,通常将受试制剂在机体内的暴露情况与参比制剂进行比较。 在上述定义的基础上,以药动学参数为终点评价指标的生物等
效性研究又可表述为:通过测定可获得的生物基质(如血液、血浆、血清)中的药物浓度,取得药代动力学参数作为终点指标,藉此反映药物释放并被吸收进入循环系统的速度和程度。通常采用药代动力学终点指标C max和AUC进行评价。 如果血液、血浆、血清等生物基质中的目标物质难以测定,也可通过测定尿液中的药物浓度进行生物等效性研究。 药效动力学研究: 在药动学研究方法不适用的情况下,可采用经过验证的药效动力学研究方法进行生物等效性研究。 临床研究: 当上述方法均不适用时,可采用以患者临床疗效为终点评价指标的临床研究方法验证等效性。 体外研究: 体外研究仅适用于特殊情况,例如在肠道内结合胆汁酸的药物等。对于进入循环系统起效的药物,不推荐采用体外研究的方法评价等效性。 二、基本要求 (一)研究总体设计 根据药物特点,可选用1)两制剂、单次给药、交叉试验设计;2)两制剂、单次给药、平行试验设计;3)重复试验设计。 对于一般药物,推荐选用第1种试验设计,纳入健康志愿者参与研究,每位受试者依照随机顺序接受受试制剂和参比制剂。对于
罗红霉素胶囊说明书 【药品名称】 通用名:罗红霉素胶囊 英文名:Roxithromycin Dispersible Capsules 汉语拼音:Luohongmeisu Jiaonang 本品主要成分为罗红霉素, 化学名为9—[O—[(2-甲氧基乙氧基)-甲基]肟]红霉素。 分子式:C41H76N2O15 分子量:837.03 【性状】本品为胶囊剂。 【药理毒理】本品为半合成的14元环大环内酯类抗生素。抗菌谱与抗菌作用基本上与红霉素相仿,对革兰阳性菌的作用较红霉素略差,对嗜肺军团菌的作用较红霉素强。对肺炎衣原体、肺炎支原体、溶脲脲原体的抗微生物作用与红霉素相仿或略强。本品可透过细菌细胞膜,在接近供体(“P”位)与细菌核糖体的50S亚基成可逆性结合,阻断了转移核糖核酸(t-RNA)结合至“P”位上,同时也阻断了多肽链自受位(“A”位)至“P”位的转移,因而细菌蛋白质合成受到抑制。 【药代动力学】口服吸收好,血药峰浓度(Cmax)高,单剂量口服罗红霉素150 mg后约2小时达血药峰浓度(Cmax)6.6~7.9 mg/L,进食可使生物利用度下降约一半。分布广,扁桃体、鼻窦、中耳、肺、痰、前列腺及其他泌尿生殖道组织中的药物浓度均可达有效治疗水平。其蛋白结合率在血浓度2.5 mg/L时为96%。以原形及5个代谢物从体内排出,7.4%自尿液排出。血消除半衰期(t1/2?)为8.4~15.5小时。 【适应症】本品适用于化脓性链球菌引起的咽炎及扁桃体炎,敏感菌所致的鼻窦炎、中耳炎、急性支气管炎、慢性支气管炎急性发作,肺炎支原体或肺炎衣原体所致的肺炎;沙眼衣原体引起的尿道炎和宫颈炎;敏感细菌引起的皮肤软组织感染。 【用法用量】 空腹口服,一般疗程为5~12日。 成人一次150mg,一日2次;也可一次300mg,一日1次。 儿童一次按体重2.5~5mg/kg,一日2次。 【不良反应】主要不良反应为腹痛、腹泻、恶心、呕吐等胃肠道反应,但发生率明显低于红霉素。偶见皮疹、皮肤瘙痒、头昏、头痛、肝功能异常(ALT及AST升高)、外周血细胞下降等。 【禁忌症】对本品、红霉素或其他大环内酯类药物过敏者禁用。 【注意事项】 1.肝功能不全者慎用。严重肝硬化者的血消除半衰期(t1/2?)延长至正常水平2倍以 上,如确实需要使用,则一次给药150mg,一日1次。 2.轻度肾功能不全者不需作剂量调整,严重肾功能不全者给药时间延长一倍(一次 给药150mg,一日1次)。 3.本品与红霉素存在交叉耐药性。 4.为获得较高血药浓度,本品需空腹(餐前1小时或餐后3~4小时)与水同服。 5.用药期间定期随访肝功能。
USP<1090>体内生物等效性试验指南第一部分 背景 该章节提出进行药物制剂性能体内及体外评估的有关建议。该章节提供指导目的是为科学家或医师欲通过找到可以替代与人体临床试验相关或临床试验前研究的方法,用于评估药物制剂性能。USP-NF提供原料药、辅料以及成品的质量标准。法定物质或制剂的USP-NF中每一个品种正文均对应一个官方批准的原料药或制剂。品种正文包括产品定义;包装,储存条件;以及质量标准内容。质量标准包括一系列通用的检查(性状、鉴别、杂质、含量测定)以及特定的检查项目,每个检查具有一个或多个分析方法以及限度要求。质量标准是药物制剂不可或缺的重要属性。满足USP-NF的标准,在全球范围内均可作为高质量药物制剂的保障,并且是生物等效性(BE)、可替代的多来源药物制剂获批的必要要求。多来源药物制剂(Multisource drugproducts)必须达到体内及/或体外试验特性标准,以确认具有治疗等效性及可替代性。在不同的国家,可替代的多来源药物制剂的法规获批情况是不同的(参考即将颁布的章节《药物制剂选择的要点Essentials for DrugProduct Selection》(1096)仍在议)。药物制剂(drug performance)可被定义为活性成分(API)从药物制剂中的释放,产生API的体内利用度可以获得理想的疗效。该章节讨论了决定药物制剂特性的体内及体外方法,重点讨论口服固体制剂方面。 该章节参考了FDA指导原则,《行业指导原则-口服制剂生物利用度及生物等效性研究-基本研究Guidance forIndustry—Bioavailability and Bioequivalence Studies for Orally AdministeredDrug Products—General Considerations(2003)》(https://www.doczj.com/doc/3417872083.html,/;请以文件名检索),以及WHO文件,《附录7 多来源(仿制)药物制剂:建立可替代性注册要求的指导原则Annex7 Multisource (Generic) Pharmaceutical Products: Guidelines on RegistrationRequirements to Establish Interchangeability(2006)》(http://who.int/en/;请以文件名检索)。FDA 的指导原则主要适合于在美国境内使用;各国家/地区药物制剂监督机构可以使用WHO、FDA和其他由国家/地区的指南。一旦获得批准,药物制剂质量控制可以在一定程度上由内部或公开标准进行,包括性能检查。USP提供了以下通则,描述了这些检查及实施步骤:《崩解度》<701>、《溶出度》<711>、《药物释放》<724>,《药物制剂的体内及体外评价》<1088>以及《溶出度实验方法的建立和验证》(1092)。 该章节提供了生物等效性(BE)研究以及溶出曲线对比实施的通用信息,BE研究作为体内药物制剂特性检查的替代方法,溶出曲线对比作为体外药物制剂特性检查的方法。该章节还讨论了针对特定药物制剂的体内BE豁免条件,并且叙述了如何应用生物药剂学分类系统(BCS)进行药物制剂性能的预测。该章节的附件定义了关键科学术语,并提供了FDA及WHO药物制剂释义评估的对比。 生物利用度,生物等效性以及溶出度 生物利用度(BA)研究主要是测定药物从口服制剂中释放并到达作用部位的过程及速度(参见FDA指导原则《行业指导原则-口服制剂生物利用度及生物等效性研究-基本研究Guidancefor Industry—Bioavailability and
人体生物利用度及 生物等效性研究
药物代谢与药动学实验室 2012.11.2 王雪丁
生物利用度(BIOAVAILABILITY )
| 概念:指药物从某制剂吸收进入全身血循环
的速度和程度
| 意义:评价药物制剂质量的重要指标,也是
选择给药途径的依据之一
| 分类
绝对生物利用度(F) 相对生物利用度( 相对 物利用度 Fr)
2
绝对生物利用度(F)
| 指血管外给药后,吸收进入血循环的药物量
占所给予的药物总量(静脉给药的药物暴露 量 的 例 其表示为 量)的比例,其表示为:
绝对生物利用度(F) = AUC血管外 AUC静脉注射
3
相对生物利用度(FR)
| 指血管外途径给予的两种制剂按剂量校正后
(等剂量使用?) 二者吸收进入血循环的 (等剂量使用?),二者吸收进入血循环的 药物量之 药物量之比
受试制剂的AUC 相对生物利用度(Fr) = 参比制剂的AUC
4
二、生物利用度试验需具备的条件
生物利用度试验是在人体进行的,因此,根 据我国药品GCP的精神和规定的人体临床试验要求 进行。
5
三、生物利用度试验与临床研究的相关性
(一)生物利用度与生物等效性 物利用度与 物等效性
| 生物利用度:以药物的血药浓度及药时曲线下面
积为基础,它反映了药物吸收的程度与速度,许 多药物的疗效与毒性往往与血药浓度有关。
6
《慢性咽炎偏方方案库: 罗红霉素胶囊+咽炎片治疗方法 该方来自csdn一个哥们,他的原文为: 认识我的人都知道,我酷爱辣椒,而且吃的量也比较大,比如2003年去长沙那次,在中南大学对面的一个面馆,湖南特有的那种装辣椒的罐子,我一碗放了它三分之一以上将近一半的剁椒到碗里。 所以,我的慢性咽炎到我治好的那一年已经差不多二十年了,很多朋友也都知道我平时咳嗽的比较多,呵呵。 今天写这个是因为看到gmail的一个消息说的是慢性咽炎的治疗,我想拿我自己的经历告诉大家不要花费冤枉钱了。 电视上最近打广告比较厉害的那个治疗慢性咽炎的药物,我也吃过,那是2002年时候就吃过,没有任何效果。 2004年离开成都前,在一个药房,一个四十多岁的药房大姐给我推荐了两种药,下面是我添加的连接:https://www.doczj.com/doc/3417872083.html, /qingrun/Gallery/379561.aspx 这两种药和生产厂家一定要确认,否则会无效的: 1、长春银诺克药业有限公司生产的咽炎片,饭前吃,每日三次,每次四片 2、河南天工药业生产的天诺宏牌罗红霉素胶囊,晚饭后半小时吃,每日一次,每次两粒 大概吃半年就足够了,一般吃上一个月就可以感觉到好转。 我当时因为第一次买也只买了一个多月的,吃了以后的确感觉痰少多了,但是因为在北京没有找到这两种药,所以就没买,大概半年后回成都,第二次买了三个月的,中间又停了三四个月,第三次回去又买了三个多月的药,吃完了,到现在已经一年多了就一直没有那种咽喉有痰的感觉了。 当然,如果空气环境和饮食习惯不好,可能还会有痰出现,但都没有那么难受了。 发这个非技术帖子,主要是想让大家不要浪费时间和金钱在没有疗效的药上,当然也可能我的咽炎和您的不一定一样,但是试验一下,应该没什么大问题,这些药都不是可以致命的。
生物等效性试验方法及规程 生物等效性主要包括临床应用得安全性与有效性。仿制药得研究开发与临床药品应用得替换,其基本要求都就是不同制剂间具有生物等效性。所以,生物等效性试验有着非常重要得地位与作用。但就是对于试验方法,很多都不知道,下面就为大家简单得介绍一下吧 生物等效性试验方法一般包括体内与体外两种方法,下面就为大家简单得介绍一下: 1、药代动力学法:测量生物样本如全血,血浆,血清,或其她生物样本中药物得活性成份,或其代谢产物得浓度与时间得关系;体外法:此种方法具有已确立好得体内外相关关系,可用于预测人体生物利用度得相关数据. 2、人体体内法:测量尿样样本中药物得活性成份,或其代谢产物得浓度与时间得关系。 3、药效法:测量药物得活性成份,或其代谢产物得即时药效与时间得关系。 4、临床试验法:通过设计良好得临床比较试验以综合得疗效终点指标来确立生物等效性。 5、体外方法通常为体外溶出度测定法:能够确保体内生物利用度。 6、FDA认可得任何其它用于测量生物利用度与生物等效性得方法。 以上就是我为大家介绍得一些方法,现在就来简单得介绍一下实验前应准备那些: 1、材料 1、1药政部门同意进行生物等效性试验得批文,同一批号得药检部门得检验报告书。 1、2同类制剂得临床文献,应有疗效分析,不良反应及药代动力学得内容。 1、3受试药得临床前药理与毒理试验得报告及生物等效性试验得计划。 1、4受试药制剂及少量纯品(供作标准曲线用),参比药制剂。 2、受试者 为了减少个体误差并保障受试者得安全,应注意以下几点:
2、1选男性青年:年龄相差不超过10岁。身长以160一180cm为宜。体重应在标准体重土10%范围内。我国标准体重可按下式估算:标准体重kg二0、7火(身高cm一8得。特殊药物可选用妇女、儿童、肿瘤病人,不受上述限制。 2、2受试前检查:心电图、血压、肝肾功能、血常规等应正常,记录既往病史与既往用药史。注意过敏体质及有药物过敏史者切勿入选。受试2wk前未用其她药物。 2、3列表报告:体重、身长、心率、血压、ALT、BUN、Cr及用药顺序(TOR或ROT)。 2、4知情同意书:有每人病史、体检、化验结果及签名,伦理委员会总批件。 2、5例数:国内规定12一24例,FDA18一24例。例数少难合格,误差大者例数应多一 些。 2、6管理:用药前禁食12h,用药后禁食4h,统一上午7时(或8时)用药,以温开水200ml送服。实验期间统一饮食,用膳时间应一致,以免影响药峰时间。不吃茶、酒、高脂食物,不作剧烈运动或整日卧床,否则误差增大。 2、7取血点:FDA规定16-20个点,我国9-16个点。至少3个半衰期或最低药浓小于峰值药浓得1/10,建议根据文献或先以较密间距实测1-2人,以上升段、峰值段各3-4点,下降段4-6点为宜。最后一点至少已有3个半衰期或药浓小于峰值药浓得1/10-1/20。 2、8清洗期通常取1wk,说明间隔几个半衰期,就是否已达到清洗目得。 3、药物 3、1供试药及参比药均应报告药名、制剂、规格、厂商、批号、 3、2参比药应说明选用理由宜用已上市得同样产品。化学上就是同一物质,处方、生产工艺、制剂及剂量基本一致。特殊情况下可用不同规格、剂量或剂型,应充分说明理由。 3、3检测应报告仪器、试剂、内标物得规格、来源、详细检测条件(色谱柱、流动相、流速、波长、灵敏度、温度)及血药浓度检测方法。报告标准曲线(R>0,98)、回归方程、线性范围。 最低检出浓度。列出标准品、血浆及含药血浆得图谱以说明专属性及干扰情况。理论塔板数不得小于1000。回收率作3个浓度各5次重复,绝对回收率>70%,RSD<10%-15%。精密试验作3个浓度各5次重复得日间、日内差异,RSD(15%-20%)。 4、药代动力学
附件3 仿制药质量一致性评价人体生物等效性研究 技术指导原则 一、概述药物制剂要产生最佳疗效,其药物活性成分应当在预期时间段内释放吸收并被转运到作用部位达到预期的有效浓度。大多数药物是进入血液循环后产生全身治疗效果的,作用部位的药物浓度和血液中药物浓度存在一定的比例关系,因此可以通过测定血液循环中的药物浓度来获得反映药物体内吸收程度和速度的主要药代动力学参数,间接预测药物制剂的临床治疗效果,以评价制剂的质量。允许这种预测的前提是制剂中活性成分进入体内的行为是一致并且可重现的。 生物利用度 (Bioavailability,BA )是反映药物活性成分吸 收进入体内的程度和速度的指标。过去出现的一些由于制剂生物利用度不同而导致的不良事件,使人们认识到确有必要对制剂中活性成分生物利用度的一致性或可重现性进行验证,尤其是在含有相同活性成分的仿制产品要替代它的原研制剂进入临床使用的时候。鉴于药物浓度和治疗效果相关,假设在同一受试者,相同的血药浓度- 时间曲线意味着在作用部位能达到相同的药物浓度,并产生相同的疗效,那么就可以药代动力学参数作为替代的终点指标来建立等效性,即生物等效性 ( Bioequivalence, BE)。
BA和BE研究已经成为评价制剂质量的重要手段。本指导原则 将重点阐述BA和BE研究的相关概念、应用范围和BA和BE研究的设计、操作和评价等。 本指导原则主要是针对化学药品普通固体口服制剂质量一致性评价的人体生物等效性研究。因为在具体应用过程中有可能面临多种情况,对于一些特殊问题,仍应遵循具体问题具体分析的原则。 二、BA和BE基本概念及应用 1. 生物利用度:是指药物活性成分从制剂释放吸收进入全身循环的程度和速度。一般分为绝对生物利用度和相对生物利用度。绝对生物利用度是以静脉制剂(通常认为静脉制剂生物利用度为100%)为参比制剂获得的药物活性成分吸收进入体内循环的相对量;相对生物利用度则是以其他非静脉途径给药的制剂(如片剂和口服溶液)为参比制剂获得的药物活性成分吸收进入体循环的相对量。 2. 生物等效性:是指药学等效制剂或可替换药物在相同试验条件下,服用相同剂量,其活性成分吸收程度和速度的差异无统计学意义。通常意义的BE研究是指用BA研究方法,以药代动力学参数为终点指标,根据预先确定的等效标准和限度进行的比较研究。在药代动力学方法确实不可行时, 也可以考虑以临床综合疗效、药效学指标或体外试验指标等进行比较性研究,但需充分证实所采用的方法具有科学性和可行性。 了解以下几个概念将有助于理解BA和BE 原研药(Innovator Product ):是指已经过全面的药学、 药理学和毒理学研究以及临床研究数据证实其安全有效性并首次被批准上市的药品。药学等效性(Pharmaceutical
药物制剂人体生物利用度和生物等效性试验指导原则
附录三药物制剂人体生物利用度和生物等效性试验指导原则生物利用度是指剂型中的药物被吸进入血液的速率和程度。生物等效性是指一种药物的不同制剂在相同的试验条件下,给以相同的剂量,反映其吸收速率和程度的主要动力学参数没有明显的统计学差异。 口服或其他非脉管内给药的制剂,其活性成分的吸收受多种因素的影响,包括制剂工艺、药物粒径、晶型或多晶型,处方中的赋形剂、黏合剂、崩解剂、润滑剂、包衣材料、溶剂、助悬剂等。生物利用度是保证药品内在质量的重要指标,而生物等效性则是保证含同一药物的不同制剂质量一致性的主要依据。生物利用度与生物等效性概念虽不完全相同,但试验方法基本一致。为了控制药品质量,保证药品的有效性和安全性,特制定本指导原则。何种药物制剂需要进行生物等效性或生物利用度试验,可根据有关部门颁布的法规要求进行。 进行药物制剂人体生物利用度和生物等效性试验的临床实验室和分析实验室,应提供机构名称以及医学、科学或分析负责人的姓名、职称和简历。 一、生物样品分析方法的基本要求 生物样品中药物及其代谢产物定量分析方法的专属性和灵敏度,是生物利用度和生物等效性试验成功的关键。首选色谱法,如HPLC、GC以及GC-MS、LC-MS、LC-MS-MS联用技术,一般应采用内标法定量。必要时也可采用生物学方法或生物化学方法。
由于生物样品取样量少、药物浓度低、内源性物质(如无机盐、脂质、蛋白质、代谢物)及个体差异等多种因素影响生物样品测定,所以必须根据待测物的结构、生物介质和预期的浓度范围,建立适宜的生物样品分析方法,并对方法进行验证。 1.专属性必须证明所测定的物质是原形药物或特定的活性代谢物,内源性物质和相应的代谢物不得干扰样品的测定。对于色谱法至少要提供空白生物样品色谱图、空白生物样品外加对照物质色谱图(注明浓度)及用药后的生物样品色谱图。对于复方制剂应特别加强专属性研究,以排除可能的干扰。对于LC-MS和LC-MS-MS方法,应着重考察基质效应。 2.标准曲线与线性范围根据所测定物质的浓度与响应的相关性,用回归分析方法获得标准曲线。标准曲线高低浓度范围为线性范围,在线性范围内浓度测定结果应达到试验要求的精密度和准确度。 必须用至少6个浓度建立标准曲线,应使用与待测样品相同的生物介质,线性范围要能覆盖全部待测浓度,不允许将线性范围外推求算未知样品的浓度。标准曲线不包括零点。 3.精密度与准确度要求选择3个浓度的质控样品同时进行方法的精密度和准确度考察。低浓度选择接近定量下限(LLOQ),在LLOQ的3倍以内;高浓度接近于标准曲线的上限;中间选一个浓度。每一浓度至少测定5个样品。 精密度用质控样品的日内和日间相对标准差(RSD)表示,RSD一般应小于15%,在LLOQ附近RSD应小于20%。 准确度是指用特定方法测得的生物样品浓度与真实浓度的接近程度,一般应在85%~115%范围内,在LLOQ附近应在80%~120%范围内。