
分散固相萃取 超高效液相色谱串联质谱
快速测定牛奶中19种 内酰胺类药物及其代谢物
祝伟霞,刘亚风*,袁 萍,郭俊峰,杨冀州,魏 蔚,孙转莲
(河南出入境检验检疫局,郑州450003)
摘 要:建立了同时测定牛奶中19种 内酰胺类药物及其代谢物的高通量液相色谱串联质谱(UPLC M S/M S)确证方法。以乙腈为提取溶剂,正己烷除脂后,采用C18填料分散固相萃取净化,在电喷雾正离子多反应监测模式下监测, 19种化合物在6m i n内实现分离。方法的检测限为0 2~1 g/kg;方法的定量限为1~5 g/kg;在3个添加浓度水平时平均回收率为66%~107%,相对标准偏差为5 6%~15%。该方法可用于测定牛奶中19种 内酰胺类药物及其代谢物。
关键词: 内酰胺;分散固相萃取;超高效液相串联质谱;牛奶
中图分类号:O657 63 文献标识码:A 文章编号:1000 0720(2010)07 029 05
内酰胺类抗生素是结构中具有抗菌活性基团 内酰胺环的一类抗生素,广泛用于治疗动物尿道、胃肠道和呼吸道感染等疾病。 内酰胺主要包括青霉素类与头孢菌素两类药物,结构中分别含有6 氨基青霉烷酸和7 氨基头孢烷酸的母核结构。 内酰胺类抗生素作用特点是通过共价键与粘肽转肽酶结合,阻止细菌细胞壁的合成,呈现杀菌活性[1]。
早期检测 内酰胺类抗生素常采用气相色谱测定其甲酯化衍生物;微生物法是一种成本低廉、操作简便的初筛方法,但其存在假阳性率高,特异性差等缺点;由于所有的 内酰胺类药物均在220~280nm处有吸收,缺乏特征的紫外发色基团,液相色谱柱前或柱后衍生化常用于医药和生物体液中 内酰胺类药物分析[2],免疫法和毛细管电泳法[3,4]也被用于样品中 内酰胺类药物的测定;目前国内外已有许多采用液相色谱串联质谱测定动物源性食品中 内酰胺的报道[5~8],但存在样品前处理步骤复杂、色谱分离时间长及涵盖待测物种类少等不足。
本实验通过对牛奶中多种 内酰胺类药物及其代谢物残留量测定的研究,运用简单的分散固相萃取净化方法,建立了19种 内酰胺类药物及其代谢物多残留的UPLC M S/M S分析方法。
1 实验部分
1 1 仪器和试剂
API4000Q液相色谱 串联线性离子阱质谱联用仪(美国应用生物公司),配W aters超高效液相色谱仪和电喷雾离子源;I KA T18涡旋混匀器; Ya m ato8210型水浴超声波装置;日立5810型离心机;氮气浓缩仪(OA SYS)。
标准品:阿莫西林(Am ox ic illin)、氨苄西林(Am p icilli n)、苄青霉素(penic illin G)、苯氧甲基青霉素(pen icilli n V)、双氯青霉素(d icloxacilli n)、邻氯青霉素(cloxacilli n)、奈夫西林(nafcillin)、苯唑青霉素(ox ac illi n)、头孢哌酮(Ce faperazone)、去乙酰基头孢匹林(D eacety lefapirin)、头孢洛宁(Ce falon i u m)、头孢羟氨苄(Cefadr ox il)、头孢喹咪(C efquino m e)、头孢呋辛(Ce f u rox i m e)、头孢拉定(C efradine)、头孢氨苄(C efalex in)、头孢噻呋(C eftiofur)、头孢唑啉(Cefazolin)、头孢匹林(Ce fap iri n),均购自美国S i g m a A ldrich公司和德国
*收稿日期:2009 10 18;修订日期:2009 12 26
基金项目:河南省普通科技攻关(0524430004)项目资助
作者简介:祝伟霞(1979-),女,助理工程师;E m a i:l li uy@f hac i q gov cn
Dr Ehrenstorfer公司,纯度97%);C18键合相填料(美国Ag ilent公司);甲酸(96%)和乙腈(色谱纯,美国Fisher公司);其它试剂均为分析纯;水为M illipore纯水系统制得高纯水(18M/c m)。
1 2 标准溶液的配制
分别称取按质量折算为100%的标准品10m g 于100mL容量瓶中,加入水溶解并定容至刻度,该单个标准储备溶液浓度为100 g/mL(-20!冷冻保存)。分别取上述储备液用乙腈-0 1%甲酸(1?1)稀释质量浓度为1 0 g/mL的质谱调谐溶液。分别取上述储备液用乙腈 水(1?1)配制成质量浓度为1 0 g/mL混合工作溶液。
1 3 仪器条件
色谱柱:W ater UPLC H SS T3(100mm#2 1mm
i d ,1 7 m);柱温:30!;流动相A:乙腈,B:
0 1%甲酸溶液;洗脱梯度程序:0m in(10%A)~
1 5m in(20%A),保持1m i n,
2 5m i n(20%A)~
3 5m i n(60%A)保持1 5m in,5(60%A)~
5 5m in(95%A)保持1m i n,
6 5m in(95%A)~
6 7m i n(10%A)保持2 8m i n;流速:0 3mL/ m i n;进样量:10 L;V ac l o六通阀:0~6m in?质谱(A位),6m i n以后?废液(B位)。
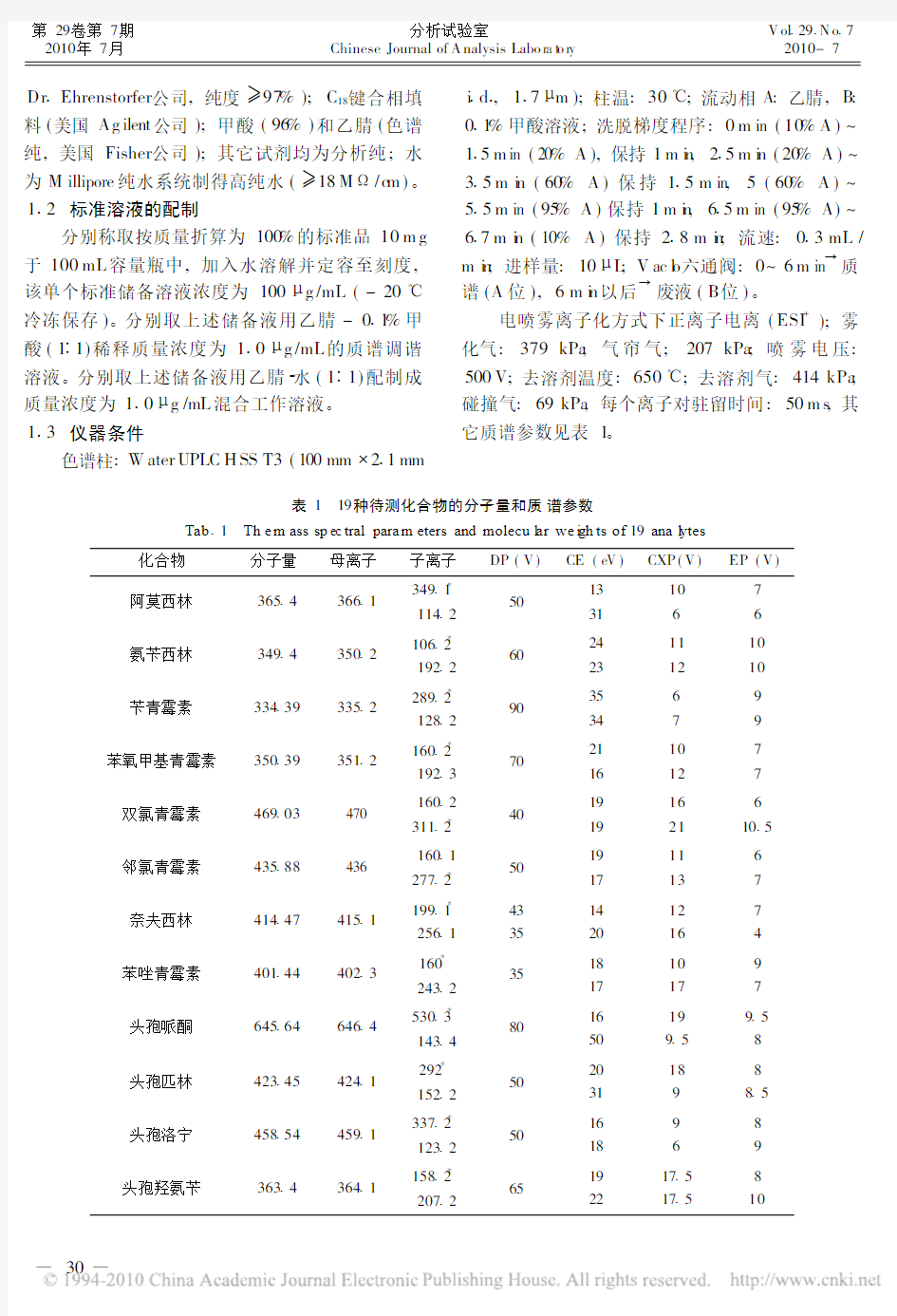
电喷雾离子化方式下正离子电离(ESI+);雾化气:379kPa;气帘气;207kPa;喷雾电压: 500V;去溶剂温度:650!;去溶剂气:414kPa;碰撞气:69kPa,每个离子对驻留时间:50m s,其它质谱参数见表1。
表1 19种待测化合物的分子量和质谱参数
Tab 1 Th e m ass sp ec tral para m eters and molecu l ar we i gh ts of19ana l ytes
化合物分子量母离子子离子DP(V)CE(eV)CXP(V)EP(V)
阿莫西林365 4366 1349 1*
114 2
50
13
31
10
6
7
6
氨苄西林349 4350 2106 2*
192 2
60
24
23
11
12
10
10
苄青霉素334 39335 2289 2*
128 2
90
35
34
6
7
9
9
苯氧甲基青霉素350 39351 2160 2*
192 3
70
21
16
10
12
7
7
双氯青霉素469 03470
160 2
311 2*
40
19
19
16
21
6
10 5
邻氯青霉素435 88436
160 1
277 2*
50
19
17
11
13
6
7
奈夫西林414 47415 1199 1*
256 1
43
35
14
20
12
16
7
4
苯唑青霉素401 44402 3160*
243 2
35
18
17
10
17
9
7
头孢哌酮645 64646 4530 3*
143 4
80
16
50
19
9 5
9 5
8
头孢匹林423 45424 1292*
152 2
50
20
31
18
9
8
8 5
头孢洛宁458 54459 1337 2*
123 2
50
16
18
9
6
8
9
头孢羟氨苄363 4364 1158 2*
207 2
65
19
22
17 5
17 5
8
10
续表1(Con ti nued T ab 1)
化合物分子量母离子子离子DP(V)CE(eV)CXP(V)EP(V)
头孢喹咪528 6529 2134 3*
396 1
55
23
19
8
10
10
7
头孢拉定349 4350 2158 2*
193 3
65
23
20
9
12
9
9
头孢呋辛424 39442 1364 1*
336 1
45
15
22
11
8
4
4
头孢氨苄347 39348 2158 2*
191 2
65
23
22
10
13
8
9
头孢噻呋523 55524 2241 2*
210 2
70
26
32
16 5
13 5
11
10
头孢唑啉454 5455 1323 2*
156 2
55
16
23
8
8
8 5
8
去乙酰基头孢匹林381 42382112*
152
36
35
27
5
16
11
11
注:*为定量离子;DP为去簇电压,CE为碰撞能量,CX P为碰撞室出口电压,EP为射入电压
1 4 样品前处理
量取5m L牛奶样品置于50mL离心管中,加入20mL乙腈,涡旋混匀1m i n,超声10m in,以4000r/m in离心5m i n,转移上清液于一带刻度50mL离心管中,再加入5m L乙腈于牛奶残渣中,涡旋混匀后离心,合并上清液并定容至30mL,加入10mL乙腈饱和的正己烷,液液分配后离心,弃去己烷层,再加入10mL乙腈饱和的正己烷重复液液萃取操作并弃去己烷层。吸取6mL上清液加至0 5g C18填料中,涡旋混匀3m in,静置5m i n 使样液充分与填料接触,以4000r/m in离心2m i n,转移上清液于另一试管中,经0 45 m滤膜过滤后氮吹浓缩至干(温度低于45!),用1mL乙腈 0 1%甲酸溶液(1?9)溶解残渣,经
0 22 m滤膜过滤,供UPLC M S/MS测定。
1 5 头孢匹林代谢物的制备
称取10m g头孢匹林标准品,加入10mL水溶解,将该溶液加至5g阴性牛肝脏样品中,涡旋混匀3m in后,于45!下温育过夜,取出加入20mL乙腈振荡提取,于4000r/m i n离心5m i n,转移上清液于100mL容量瓶中,再依次用乙腈提取、离心并转移上清液,并用乙腈定容至100mL。
2 结果与讨论
2 1 净化方法的选择
内酰胺类药物测定常用反相固相萃取柱技术,如C18或H LB小柱,因提取液中常含有少量有机溶剂,采用该方法进行SPE净化时,必须把提取液中有机溶剂完全蒸干。为简化操作,本实验选用C18和PSA填料进行分散固相萃取,实验数据表明PSA填料净化 内酰胺化合物时回收率低(20%~82%),是由于结构中的羰基与PSA填料产生离子交换作用的结果。C18填料应用于动物源性食品中19种 内酰胺类药物及其代谢物净化时,每一种待测物均获得较高回收率。
2 2 头孢匹林与其代谢物
因为在动物组织中存在酯酶,头孢匹林在动物体内易代谢为去乙酰基头孢匹林[7~9],M B ec ker报道[10]头孢匹林在25!时10m in内能完全转化为其代谢物,但在牛奶中不代谢。因此欧盟规定牛奶中头孢匹林及其代谢物总量为100 g/kg,头孢匹林残留检测中必须检测其代谢物。目前还没有商品化去乙酰基头孢匹林标准物质上市,本实验合成了去乙酰基头孢匹林,采用头孢匹林(1 0m g/mL)标准液加入牛肝脏中于37!下过夜,取出后采用本方法进行前处理,经液相色谱 二极管阵列检测后定量,结果表明去乙酰基头孢匹林反应产率为90%~98%。
2 3 流动相的选择
除阿莫西林、氨苄西林、奈夫西林、头孢拉定、头孢氨苄、头孢羟氨苄具有两性电离性质
外[8],其它 内酰胺均为弱有机酸类化合物,为了增强不同待测化合物在C18键合相中的保留能力,分别比较了乙腈/水、甲醇/水、乙腈/甲酸溶液、甲醇/甲酸溶液、乙腈/甲酸溶液/乙酸胺溶液等作为流动相时的分离效果。结果发现有机相采用甲醇时色谱峰变宽;乙酸胺缓冲液加入流动相中,19种 内酰胺灵敏度全部下降,其中头孢喹咪下降1/3。本文研究了甲酸体积分数对灵敏度和分离度的影响,比较了甲酸体积分数(0 02%、0 05%、0 1%、0 2%、0 3%、0 5%)对质谱响应及色谱分离的影响,实验发现甲酸体积分数对保留时间影响不大,但是对灵敏度影响较大,体积分数为0 1%甲酸时响应最高,最终确定乙腈 0 1%甲酸作为流动相。
2 4 质谱测定条件的优化
由于羰基和氨基存在于结构中, 内酰胺类化合物在正负离子下都能产生电离,但正离子模式下相对强度高,在正离子模式下进行母离子全扫描时,除头孢呋辛正离子模式下得到[M+NH3 +H]+,其它所有化合物均得到相应的[M+H]+峰,以[M+H]+或[M+NH3+H]+为母离子,进行子离子全扫描,根据2002/657/EEC关于确证法4个确证点的要求,选取丰度较强、干扰较小的两对子离子为定性离子、定量离子。 内酰胺类化合物的特征碎片裂解主要以 内酰胺开环、6位或7位碳 NH键断裂、COOH的丢失、中性丢失而获得[11]。
2 5 回收率和精密度
取空白牛奶样品添加3个水平混合标准溶液(1#LOQ、1 5#LOQ、2#LOQ)进行回收率试验。每个添加水平平行实验6次,平均回收率及相对标准偏差(RSD)见表2。
2 6 线性范围和方法检测限
在空白牛奶样品中添加不同浓度混合标准工作溶液,按上述方法处理后测定,分别以定量离子对的峰面积(纵坐标)对浓度(横坐标)进行线性回归计算,所得相关系数r见表2。以信噪比(S/N)3确定方法定性检出限(LOD),以S/N
10为方法定量检测限(LOQ),经计算:阿莫西林、奈夫西林和头孢呋辛LOD为1 g/kg,其它化合物LOD为0 2 g/kg;阿莫西林、奈夫西林和头孢呋辛LOQ为5 g/kg,其它化合物LOQ为1 g/kg,均低于国际通行最高限量要求。
表2 方法回收率、精密度和相关系数
T ab 2 Th e recovery,p recisi on and corre l at i on coeffic i en t of th e m ethod 化合物回收率/%RSD/%r化合物回收率/%R S D/%r 阿莫西林72~837 80 9989头孢洛宁71~94110 9993氨苄西林89~976 40 9995头孢哌酮82~107130 9992苄星青霉素81~102100 9991头孢喹咪66~919 10 9998苯氧甲基青霉素92~985 60 9999头孢拉定70~899 60 9995双氯青霉素86~1049 70 9993头孢呋辛68~848 10 9973邻氯青霉素90~976 80 9995头孢氨苄82~996 50 9996奈夫西林69~93110 9914头孢噻呋69~102150 9991苯唑青霉素79~949 40 9990头孢唑啉87~1057 20 9992头孢羟氨苄88~954 90 9992头孢匹林83~1079 80 9945去乙酰基头孢匹林78~938 50 9918
2 7 样品分析
应用本方法,收集了市售牛奶及牛奶收购站处牛奶,共50个样品进行检测,共检出阳性结果6个,其结果见表3。从表中可以看出,阿莫西林、青霉素G、氨苄西林和头孢氨苄等廉价的抗生素是主要检出的 内酰胺类药物,也是目前在医药和兽药行业中应用最多的抗生素。
表3 样品测定结果
Tab 3 Th e d eter m i nat i on results of real sa m ples
样品编号
青霉素G
w/( g/kg)
氨苄西林
w/( g/kg)
阿莫西林
w/( g/kg)
头孢氨苄
w/( g/kg)
492///
5/203//
15//16/
2759///
29///167
43//72/
参考文献
[1] M D M a razuela,S Bog ia lli A na l Ch i m A cta,2009,
645:5
[2] L K anti an,i M Fa rr ,D Barce l T rends A na l Che m,
2009,28:729[3] 白国涛,储晓刚,潘国卿等 食品科学,2008,
29(7):485
[4] 张 琦,叶能胜,谷学新等 化学通报,2009,(5):
394
[5] D M H o lstege,B Puschner,G W h itehead et al J A g i rc
Food Chem,2002,50:406
[6] E D aese l e ire,H D R uyck,R V R energhe m R ap i d
Co mm un M ass Spectro m,2000,14:1404
[7] C K F agerquist,A R L ightfield R apid Commun M ass
Spectrom,2003,17:660
[8] S R i edike r,A Ry tz,R H Stadler Ana l Che m,2001,
73:1614
[9] A M W illi am,H K R aida J AOAC Int,1995,78:49
[10] M Becker,E Z ittlau,P M ichae l A na lChm i A cta,2004,
520:19
[11] K M astovs ka,A R L i ghtfield J Chro m atogr A,2008,
1202:118
Fast deter m i n ation of19 lact am s and itsm et abolites inm ilk by ultra perfor m ance li q ui d chrom atogra phy t ande m m ass spectro m e t ry and dispersion solid phase extracti o n
Z H U W ei x ia,LI U Ya feng*,YUAN P ing,GUO Jun feng,YANG J i zhou,WEI W ei and SUN Zhuan lian (H enan Entry ex it I nspecti o n and Q uaranti n e Bureau,Zhengzhou450003),Fenx i Shiyansh,i2010,29(7): 29~33
Abst ract:A h i g h t h roughput con fir m ati v e u ltra perfor m ance li q u i d chro m atog raphy tande m m ass spectro m etric (UPLC M S M S)m ethod w as deve l o ped to si m ultaneousl y de ter m ine19 lacta m s and its m etabo lites Aceton i trile w as used as an extraction so lvent A fter re m ov i n g li p so l u b ile substance w ith hexane,C18dispersi o n so li d phase ex traction(DSPE)w as introduced to c leanup pr ocedures The19ana l y tes w ere m easured i n the positive electr ospray i o nization and reco r ded w ith mu lti p le reaction m on itor i n g w ithin6m i n The li m its of detection are 0 2~1 g/kg and the li m its of quantification are1~5 g/kg,respectively The recoveries ranged fro m66%to 107%and t h e relati v e standard dev iations fro m5 6%to15%,sp i k ed w ith t h ree levels By deter m i n i n g real sa mp les,it is proved t h at the m ethod is si m ple,fas,t sensiti v e and accurate for the deter m i n ation of19 lacta m s and its m etabo lites i n m ilk
K eywords: lacta m s;DSPE;UPLC M S/M S;M ilk
消毒产品中丙酸氯倍他索和盐酸左氧氟沙星测定?液相色谱-串联质谱法 Determination of clobetasol propionate and levofloxacin hydrochloride in disinfection product - LC-MS-MS method 1 范围 本方法规定了膏霜类消毒产品中丙酸氯倍他索和盐酸左氧氟沙星残留量液相色谱-串联质谱测定方法。 本方法适用于膏霜类消毒产品中丙酸氯倍他索和盐酸左氧氟沙星残留量的测定。 取样量为0.1g时,本方法对丙酸氯倍他索和盐酸左氧氟沙星的检出限见表1。 表1 丙酸氯倍他索和盐酸左氧氟沙星的检出限、保留时间和特征离子 中文名称英文名称 检出限 (μg/g) 保留时 间(min) 特征离子(m/z) 丙酸氯倍他索Clobetasol propionate 0.009 7.83 467.0/355.2/373.4 盐酸左氧氟沙星Levofloxacin hydrochloride 0.06 1.11 362.0/260.9/318.2 2 规范性引用文件 3 原理 试样中丙酸氯倍他索和盐酸左氧氟沙星用甲醇提取,提取液经0.45μm滤膜过滤,用C18柱分离后,用液相色谱-串联质谱仪测定,正离子扫描,离子对定性,峰面积定量。 4 试剂和材料 除另有说明外,所用试剂均为分析纯,水为不含有机物的纯水,纯水中干扰物的浓度需低于方法中待测物的检出限。 4.1甲醇:农药残留级。 4.2乙腈:农药残留级。 4.3甲酸:分析纯。
4.4标准品:丙酸氯倍他索和盐酸左氧氟沙星均购自中国药品生物制品检定所,纯度≥99.8%。 4.5标准溶液:准确称取丙酸氯倍他索适量,用乙腈-水(1:1)配制成100μg/mL 的标准贮备液。准确称取盐酸左氧氟沙星适量,用纯水配制成100μg/mL的标准贮备液。准确量取上述标准贮备溶液适量,用乙腈稀释配制成浓度为10.0μg/mL 的混合标准中间溶液,将标准中间溶液转移到安瓿瓶中于4 C保存。临用前,再根据需要用甲醇配制成不同浓度的标准使用溶液。 4.6甲酸溶液(0.2%,v/v):量取2mL甲酸,用纯水定容至1000mL。 4.7 0.45μm滤膜。 5 仪器 5.1 液相色谱-串联质谱联用仪:HP1100高效液相色谱仪(Agilent) - API 4000质谱仪(Applied Biosystems) ,电喷雾离子化源(ESIMS,NI/PI模式)。 5.2 分析天平:感量0.1mg和0.001g。 5.3实验室纯水机:Barnstead纯水机。 5.4涡旋振荡器:Scientific Industries 涡旋振荡器。 5.5 具塞试管:10mL。 6 试样的制备与保存 6.1 试样的制备 取有代表性样品5g,搅拌均匀,制成实验室样品。 6.2 试样保存 制备好的试样置于室温保存。 7 测定步骤 7.1样品前处理 称取0.1g~0.2g样品(精确到0.001 g) ,置于10mL试管中,加入3.00mL甲醇溶液,涡旋振摇使样品分散后,超声振荡10min。静置,吸取上清液经滤膜(4.7)过滤后,供液相色谱-串联质谱测定。
气相色谱法质谱联用 气相色谱法–质谱法联用(英语:–,简称气质联用,英文缩写)是一种结合气相色谱和质谱地特性,在试样中鉴别不同物质地方法.地使用包括药物检测(主要用于监督药物地滥用)、火灾调查、环境分析、爆炸调查和未知样品地测定.也用于为保障机场安全测定行李和人体中地物质.另外,还可以用于识别物质中以前认为在未被识别前就已经蜕变了地痕量元素. 已经被广泛地誉为司法学物质鉴定地金标方法,因为它被用于进行“专一性测试”.所谓“专一性测试”就是能十分肯定地在一个给定地试样中识别出某个物质地实际存在.而非专一性测试则只能指出试样中有哪类物质存在.尽管非专一性测试能够用统计地方法提示该物质具体是那种物质,但存在识别上地正偏差. 目录 历史 仪器设备 吹扫和捕集 质谱检测器地类型 分析 全程扫描 选择地离子检测 离子化类型 电子离子化 化学离子化 串联 应用 环境检测和清洁 刑事鉴识 执法方面地应用 运动反兴奋剂分析 社会安全 食品、饮料和香水分析
天体化学 医药 参考文献 参考书目 外部链接 历史用质谱仪作为气相色谱地检测器是上个世纪年代期间由和首先开发地.当时所使用地敏感地质谱仪体积庞大、容易损坏只能作为固定地实验室装置使用. 价格适中且小型化地电脑地开发为这一仪器使用地简单化提供了帮助,并且,大大地改善了分析样品所花地时间.年,美国电子联合公司(, . 简称)美国模拟计算机供应商地先驱在开始开发电脑控制地四极杆质谱仪. 地指导下[]开始开发电脑控制地四极杆质谱仪.到了年,和地分部合作售出多台四极杆残留气体分析仪.年,仪器公司(,简称)组建就绪,年初就给斯坦福大学和普渡大学发送了第一台地最早雏型.最后重新命名为菲尼根公司()并且继续持世界系统研发、生产之牛耳. 年,当时最尖端地高速()单元在不到秒地时间里,完成了火灾助燃物地分析,然而,如果使用第一代至少需要分钟.到年使用四极杆技术地电脑化地仪器已经化学研究和有机物分析地必不可少地仪器.今天电脑化地仪器被广泛地用在水、空气、土壤等地环境检测中;同时也用于农业调控、食品安全、以及医药产品地发现和生产中. 气质联用色谱是由两个主要部分组成:即气相色谱部分和质谱部分.气相色谱使用毛细管柱,其关键参数是柱地尺寸(长度、直径、液膜厚度)以及固定相性质(例如,%苯基聚硅氧烷).当试样流经柱子时,根据个组分分子地化学性质地差异而得到分离.分子被柱子所保留,然后,在不同时间(叫做保留时间)流出柱子.流出柱子地分子被下游地质谱分析器做俘获,离子化、加速、偏向、最终分别测定离子化地分子.质谱仪是通过把每个分子断裂成离子化碎片并通过其质荷比来进行测定地. 把气相色谱和质谱这两部分放在一起使用要比单独使用那一部分对物质地识别都会精细很多很多倍.单用气相色谱或质谱是不可能精确地识别一种特定地分子地.通常,经质谱仪处理地需要是非常纯地样品,而使用传统地检测器地气相色谱(如,火焰离子化检测器)当有多种分子通过色谱柱地时间一样时(即具有相同地保留时间)不能予以区分,这样会导致两种或多种分子在同一时间流出柱子.在单独使用质谱检测器时,也会出现样式相似地离子化碎片.将这两种方法结合起来则能减少误差地可能性,因为两种分子同时具有相同地色谱行为和质谱行为实属非常罕见.因而,当一张分子识别质谱图出现在某一特定地分析地保留
---------------------------------------------------------------最新资料推荐------------------------------------------------------ 高效液相色谱质谱联用HPLC .液相色谱-质谱联用技术(LC-MS)的各种模式探索一、实验目的1、了解 LC-MS 的主要构造和基本原理; 2、学习 LC-MS 的基本操作方法; 3、掌握 LC-MS 的六种操作模式的特点及应用。 二、实验原理 1、液质基本原理及模式介绍液相色谱 - 质谱法( Liquid Chromatography/Mass Spectrometry , LCMS)将应用范围极广的分离方法——液相色谱法与灵敏、专属、能提供分子量和结构信息的质谱法结合起来,必然成为一种重要的现代分离分析技术。 但是,LC 是液相分离技术,而 MS 是在真空条件下工作的方法,因而难以相互匹配。 LC-MS 经过了约 30 年的发展,直至采用了大气压离子化技术(Atmospheric pressure ionization,API)之后,才发展成为可常规应用的重要分离分析方法。 现在,在生物、医药、化工、农业和环境等各个领域中均得到了广泛的应用,在组合化学、蛋白质组学和代谢组学的研究工作中,LC-MS 已经成为最重要研究方法之一。 质谱仪作为整套仪器中最重要的部分,其常规分析模式有全扫描模式(Scan)、选择离子监测模式(SIM)。 (一)全扫描模式方式(Scan):最常用的扫描方式之一,扫描的质量范围覆盖被测化合物的分子离子和碎片离子的质量,得到的是 1/ 13
顶空固相微萃取-气相色谱-质谱联用 分析纺织品中挥发性有机物* 蔡积进张卓旻李攻科 中山大学化学与化学工程学院,广东,广州 510275 摘要本文以顶空固相微萃取(Head Space Solid Phase Microextraction,HSSPME)和 气相色谱-质谱(GC/MS)联用技术分析纺织品中的五种常见挥发性有机物(Volatile Organic Compounds,VOCs):甲苯、4-乙烯基环己烯、苯乙烯、萘和1-苯基环己烯。 优化了顶空体积、平衡时间、萃取时间、萃取温度、搅拌速率、加盐种类和浓度以及GC/MS条件。建立了快速测定纺织品中VOCs的方法,方法对五种待测物质均具有较宽线性范围,分别为0.087~870,3.32~3320,2.28~2280,0.015~150和0.5~500 ng/g;检出限分别为0.005、0.042、0.67、0.008和0.011 ng/g。分析加标实际样品,回收率在80.1~122%之间,RSD在0.8~8.6%之间。方法符合纺织品中痕量VOCs 的快速分析要求。 关键词:固相微萃取;气相色谱-质谱;纺织品;挥发性有机物 生态纺织品标准100(Oeko-Tex Standard 100)[1]是纺织品领域通行的技术标准,严格规定了残留有毒、有害VOCs的释放量。为推动纺织品质量达到出口标准,需建立有效快速的VOCs 检测方法。由于纺织品VOCs的含量很低,常规的预富集浓缩方法很难满足分析需要,达不到相应的灵敏度要求。SPME是八十年代末Pawliszyn等[2]研制开发的一种非溶剂分析萃取技术,具有操作简单、萃取速度快、选择性和适应性好等优点。而HSSPME应用于纺织品中,一方面继承了顶空技术操作简单、不受样品基体干扰的优点;另一方面又能在采样的同时进行浓缩,大大提高了分析灵敏度。国内已有学者用SPME技术对纺织品中残留干洗溶剂(如四氯乙烯和三氯乙烯等)和驱虫剂(如二氯苯和萘等)进行分析[3~5]。本文建立了HSSPME-GC-MS联用分析纺织品中常见VOCs的分析方法,方法灵敏度高,重现性好,适合于纺织品中多种痕量挥发性有机物的分析。 1 实验 1.1 仪器及操作条件 1.1.1 仪器 SPME手动取样装置,100 μm聚二甲基硅氧烷(PDMS),电磁搅拌/加热操作台,搅拌子(3.0 mm×10.0 mm),10、15、40 mL顶端带有孔盖子和聚四氟乙烯隔垫的样品瓶(Supelco 公司)。HP-6890气相色谱仪带质谱检测(MSD-5973)配G1701B.02.05工作站(Hewlett-Packard, USA),所用色谱柱为HP-VOC熔融毛细管柱(60 m×0.32 mm×1.8 μm)。 1.1.2 GC-MS的操作条件 色谱条件:进样口温度为250 ℃,进样口关闭五分钟,不分流进样。采用程序升温,初始 资金项目:国家质检总局科研资助项目(2002IK034)、 中山大学化学院第四届创新化学实验与研究基金(批准号:03002号)。 第一作者:蔡积进(1982年出生),男,中山大学化学与化工学院材料化学专业00级 指导教师:李攻科,E-mail :cesgkl@https://www.doczj.com/doc/2b5486429.html,.
2015 年版药典高效液相色谱法、质谱法
2015 版药典 --- 高效液相色谱法、质谱法 0512 高效液相色谱法 高效液相色谱法系采用高压输液泵将规定的流动相泵入装有填充剂的色谱柱,对供试品进行分离测定的色谱方法。 注入的供试品,由流动相带入色谱柱内,各组分在柱内被分离,并进入检测器检测,由积分仪或数据处理系统记录和处 理色谱信号。 1.对仪器的一般要求和色谱条件 高效液相色谱仪由高压输液泵、进样器、色谱柱、检测器、积分仪或数据处理系统组成。色谱柱内径一般为 3.9 ~ 4.6mm,填充剂粒径为 3~lOμm。超高效液相色谱仪是适应小粒径(约 2μm)填充剂的耐超高压、小进样量、低死体积、高灵敏度检测的高效液相色谱仪。 (1)色谱柱 反相色谱柱:以键合非极性基团的载体为填充剂填充而成的色谱柱。常见的载体有硅胶、聚合物复合硅胶和聚合物 等;常用的填充剂有十八烷基硅烷键合硅胶、辛基硅烷键合硅胶和苯基键合硅胶等。 正相色谱柱:用硅胶填充剂,或键合极性基团的硅胶填充而成的色谱柱。常见的填充剂有硅胶、氨基键合硅胶和氰 基键合硅胶等。氨基键合硅胶和氰基键合硅胶也可用作反相色谱。 离子交换色谱柱:用离子交换填充剂填充而成的色谱柱。有阳离子交换色谱柱和阴离子交换色谱柱。 手性分离色谱柱:用手性填充剂填充而成的色谱柱。 色谱柱的内径与长度,填充剂的形状、粒径与粒径分布、孔径、表面积、键合基团的表面覆盖度、载体表面基团残 留量,填充的致密与均匀程度等均影响色谱柱的性能,应根据被分离物质的性质来选择合适的色谱柱。 温度会影响分离效果,品种正文中未指明色谱柱温度时系指室温,应注意室温变化的影响。为改善分离效果可适当 提高色谱柱的温度,但一般不宜超过 60℃。 残余硅羟基未封闭的硅胶色谱柱,流动相 pH 值一般应在 2~8 之间。残余硅羟基已封闭的硅胶、聚合物复合硅胶或聚 合物色谱柱可耐受更广泛 pH值的流动相,适合于 pH 值小于 2 或大于 8 的流动相。 (2)检测器最常用的检测器为紫外 - 可见分光检测器,包括二极管阵列检测器,其他常见的检测器有荧光检测器、 蒸发光散射检测器、示差折光检测器、电化学检测器和质谱检测器等。 紫外- 可见分光检测器、荧光检测器、电化学检测器为选择性检测器,其响应值不仅与被测物质的量有关,还与 其结构有关;蒸发光散射检测器和示差折光检测器为通用检测器,对所有物质均有响应,结构相似的物质在蒸发光散射 检测器的响应值几乎仅与被测物质的量有关。 紫外 - 可见分光检测器、荧光检测器、电化学检测器和示差折光检测器的响应值与被测物质的量在一定范围内呈 线性关系,但蒸发光散射检测器的响应值与被测物质的量通常呈指数关系,一般需经对数转换。 不同的检测器,对流动相的要求不同。紫外 - 可见分光检测器所用流动相应符合紫外 - 可见分光光度法(通则 0401)项下对溶剂的要求;采用低波长检测时,还应考虑有机溶剂的截止使用波长,并选用色谱级有机溶剂。蒸发光散射检测 器和质谱检测器不得使用含不挥发性盐的流动相。 (3)流动相反相色谱系统的流动相常用甲醇 - 水系统和乙腈 - 水系统,用紫外末端波长检测时,宜选用乙腈 - 水系统。流动相中应尽可能不用缓冲盐,如需用时,应尽可能使用低浓度缓冲盐。用十八烷基硅烷键合硅胶色谱柱时,流动 相中有机溶剂一般不低于 5%,否则易导致柱效下降、色谱系统不稳定。
一、开机 water 2695/micromass zq4000: 开机步骤 1. 分别打开质谱、液相色谱和计算机电源,此时质谱主机内置的CPU会通过网线与计算机主机建立通讯联系,这个时间大约需要1至2分钟。 2. 等液相色谱通过自检后,进入Idle状态,依照液相色谱操作程序,依次进行操作。(具体根据液相色谱不同型号来执行,下面以2695为例)。 a.打开脱气机 (Degasser On)。 b.湿灌注(Wet Prime)。 c.Purge Injector。 d.平衡色谱柱。 3.双击桌面上的 MassLynx 4.0图标进入质谱软件。 4.检查机械泵的油的状态(每星期),如果发现浑浊、缺油等状况,或者已经累积运行超过3000小时,请及时更换机械泵油。 5.点击质谱调谐图标(MS Tune)进入质谱调谐窗口。 6.选择菜单“Options –Pump”,这时机械泵将开始工作,同时分子涡轮泵会开始抽真空。几分钟后,ZQ就会达到真空要求,ZQ前面板右上角的状态灯“Vacuum”将变绿。 7.点击真空状态图标,检查真空规的状态,以确认真空达到要求。 8. 确认氮气气源输出已经打开,气体输出压力为90 psi。 9.设置源温度(Source Temp)到目标温度。 关机 1.点击质谱调谐图标进入调谐窗口。 2.点击Standby 让MS 进入待机状态时,这时状态灯会由绿变红,这一过程是关质谱高电压的过程。 3.停止液相色谱流速,如果还需要冲洗色谱柱,可以将液相色谱管路从质谱移开到废液瓶。4.等脱溶剂气温度(ESI)或APCI探头温度降到常温,点击气体图标关闭氮气。 5.逆时针方向拧开机械泵上的Gas Ballast 阀,运行20分钟后关闭(镇气)。 a) 对于ESI源,至少每星期做一次。 b) 对于APCI源,每天做一次。 6.再次确认机械泵的Ballast阀是否已经关闭。 7.选择Option / Vent,这时质谱开始泄真空,ZQ 前面板的状态灯“Vacuum”开始闪烁,几分钟后机械泵会停止运行,这时可以关闭质谱电源。 FINNIGEN DECA 开关机及校正流程—— 1开机前准备事项 (1)确保质谱总电源开关(白色开关)及主板电源开关(黑色开关)处于关闭状态(O); (2)检查真空泵油液面,确保泵内油页面处于标定的上下两线之间; (3)查看离子源洁净程度,ESI源查看喷口是否有固体析出,毛细管口是否完好;APCI喷口是否有积液; (4)气体压力,打开高纯氮气钢瓶总阀,调节出口压力调至0.65MPa,打开高纯氦气钢瓶总阀,调节出口压力调至0.25Mpa; (5)检查壳气及辅助气接口连接紧固,松开液相管路与离子源的接口; (6)开启动力电源,电压稳定,正常;
液相色谱-质谱联用(LC-MS) LCMS分别的含义是:L液相C色谱M质谱S分离(友情赠送:G是气相^_^) LC-MS/MS就是液相色谱质谱/质谱联用 MS/MS是质谱-质谱联用(通常我们称为串联质谱,二维质谱法,序贯质谱等) LC-MS/MS与LC-MS比较,M(质谱)分离的步骤是串联的,不是单一的。 色谱法也叫层析法,它是一种高效能的物理分离技术,将它用于分析化学并配合适当的检测手段,就成为色谱分析法。 色谱法的最早应用是用于分离植物色素,其方法是这样的:在一玻璃管中放入碳酸钙,将含有植物色素(植物叶的提取液)的石油醚倒入管中。此时,玻璃管的上端立即出现几种颜色的混合谱带。然后用纯石油醚冲洗,随着石油醚的加入,谱带不断地向下移动,并逐渐分开成几个不同颜色的谱带,继续冲洗就可分别接得各种颜色的色素,并可分别进行鉴定。色谱法也由此而得名。 现在的色谱法早已不局限于色素的分离,其方法也早已得到了极大的发展,但其分离的原理仍然是一样的。我们仍然叫它色谱分析。 一、色谱分离基本原理: 由以上方法可知,在色谱法中存在两相,一相是固定不动的,我们把它叫做固定相;另一相则不断流过固定相,我们把它叫做流动相。 色谱法的分离原理就是利用待分离的各种物质在两相中的分配系数、吸附能力等亲和能力的不同来进行分离的。 使用外力使含有样品的流动相(气体、液体)通过一固定于柱中或平板上、与流动相互不相溶的固定相表面。当流动相中携带的混合物流经固定相时,混合物中的各组分与固定相发生相互作用。 由于混合物中各组分在性质和结构上的差异,与固定相之间产生的作用力的大小、强弱不同,随着流动相的移动,混合物在两相间经过反复多次的分配平衡,使得各组分被固定相保留的时间不同,从而按一定次序由固定相中先后流出。与适当的柱后检测方法结合,实现混合物中各组分的分离与检测。 二、色谱分类方法: 色谱分析法有很多种类,从不同的角度出发可以有不同的分类方法。 从两相的状态分类:
附件 面膜类化妆品中氟轻松检测方法 (高效液相色谱-串联质谱法) 1范围 本方法规定了面膜类化妆品中氟轻松的高效液相色谱-串联质谱测定方法。 本方法适用于面膜类化妆品中氟轻松的定性定量测定。 2方法提要 面膜类化妆品用饱和氯化钠溶液分散,用乙腈从分散液中提取氟轻松,用亚铁氰化钾和乙酸锌沉淀提取液中大分子基质,经固相萃取小柱净化,用高效液相色谱仪分离,质谱检测器检测,采用保留时间和特征离子对丰度比定性,以待测物质相对应离子峰面积定量,以标准曲线法计算含量。 本方法的检出限为0.03 μg/g,定量限为0.05 μg/g。 3试剂和材料 除另有规定外,本方法所用试剂均为分析纯或以上规格,水为纯化水。 3.1甲醇:色谱纯。 3.2乙腈:色谱纯。 3.3冰醋酸:优级纯。 3.4饱和氯化钠溶液。 3.5 10%亚铁氰化钾溶液:称取115 g亚铁氰化钾K4Fe(CN)6·3H2O固体,
用水溶解定容至1000 mL。 3.6 20%乙酸锌溶液:称取239 g乙酸锌C4H6O4Zn·2H2O固体,用水溶解定容至1000 mL。 3.7Oasis HLB固相萃取小柱或相当者:60 mg,3 mL。 3.8 标准物质:氟轻松,纯度不小于99.0%;标准物质的分子式、相对分子质量、CAS登录号、化学结构图参见附录A。 3.9 标准储备液(ρ=1g/L):准确称取氟轻松标准物质(3.8)10mg,精确到0.01 mg,置于10 mL量瓶中,用甲醇溶解并定容,于-18℃下冷冻保存。 3.10 标准工作溶液:临用时,取标准储备液(3.9)适量,用乙腈稀释成0.05μg/mL、0.10μg/mL、0.20μg/mL、0.40μg/mL、0.80μg/mL系列浓度的标准工作溶液。 4仪器和设备 4.1 高效液相色谱-三重四极杆质谱联用仪(ESI源)。 4.2 分析天平:感量0.0001g;0.00001g。 4.3 涡旋混合器。 4.4离心机:转速5000r/min,容量10mL;50mL。 4.5 固相萃取装置。 5分析步骤 5.1样品处理 5.1.1提取 称取样品(带有载体的面膜,去除载体后取样)0.2 g,精确至0.0001 g,置15 mL具塞离心管中,加入3 mL饱和氯化钠溶液(3.4),于涡旋混合器上混合使样品分散,准确加入2 mL乙腈,充分涡旋提取2 min,以
高效液相色谱质谱联用技术的应用 高效液相色谱(HPLC或LC)是以液体溶剂作为流动相的色谱技术,一般在室温下操作,可以直接分析不挥发性化合物、极性化合物和大分子化合物(包括蛋白、多肽、多糖、多聚物等),分析范围广,而且不需衍生化步骤。质谱是强有力的结构解析工具,能为结构定性提供较多的信息,是理想的色谱检测器,不仅特异,而且具有极高的检测灵敏度。串联质谱(MS/MS)是将一个质量选择的操作接到另一个质量选择的后面,在单极质谱给出化合物相对分子量的信息后,对准分子离子进行多极裂解,进而获得丰富的化合物碎片信息,确认目标化合物,对目标化合物定量等。[1] 高效液相色谱一质谱(HPLC—MS)联用技术是近几年来发展起来的一项新的分离分析技术,将HPLC 对复杂样品的高分离能力,与MS具有高选择性、高灵敏度及能够提供相对分子质量与结构信息的优点结合起来,在药物分析、环境分析等许多领域得到了广泛的应用。[2] 本文着重讲述液相色谱质谱联用仪在药物分析、环境分析上的应用。 1液相色谱质谱联用在药学分析上的应用 1.1LC/MS在药物代谢中的应用 Lee等[3]总结了利用LC/MS鉴定药物代谢产物的方法,主要包括以下几个步骤:测定原形药物的质谱;选择准分子离子、加合离子和主要的碎片离子进行多级质谱分析;选择原形药物的主要中性丢失,测定生物样品的中性丢失谱,图谱中的离子即为原形药物和可能的代谢物的分子离子;选择主要的子离子测定生物样品的母离子谱,所得母离子即为各个代谢物;测定生物样品中所有可能代谢物的子离子谱,解谱得到代谢物的结构。 王宁生等[4]以LC/MS联用技术及标准品对照法,分离检测健康志愿者口服复方丹参滴丸后,血清中水溶性成分及代谢产物,从一级质谱的分子离子峰推测,丹参素及原儿茶醛在体内分别与硫酸及葡萄糖醛酸结合,产生丹参素硫酸结合物及原儿茶醛的葡糖醛酸结合物。 Hsiu SL等[5]研究芍药苷在小鼠体内药代动力学,用LC/MS方法检测体内药物浓度,未检测到芍药苷原形药物;但在血浆及各种排泄物中,均可检测其代谢物,经液相色谱一质谱分析,结合核磁共振(NMR),确定其为芍药苷的脱糖基代谢物,提示芍药苷给药后,在肠道经细菌转化为PG后,被吸收进入血液循环中发挥作用。 Chen SJ等[6]用LC/DAD/MS/MS联用技术,对山豆根碱在小鼠体内的代谢进行了研究,用ESI /MSn技术检测山豆根碱的代谢物,并鉴定其主要代谢物为N一去甲基山豆根碱。 1.2LC/MS在药学浓度上的应用 M.Brolis等[7]采用I-IPLC—DAD—MS法从贯叶金丝桃Hyoericum performm中分离鉴定出槲皮素、异槲皮素、金丝桃苷等成分。 Gerthard Brillgma等[8]采用HPLC—NMR和HPLC—ESI—MS—MS法对Habropetalum dawei进行分析,分离鉴定出dioneopeltine、N-methyldioncophylline、N-methyl-7-epi-dioncophylline、tetralone、(1R,3R)和(1S,3R)-N-formyl-8-hydroxy-6-methoxy-l,3-dimthyltetra-hydroisoquinoline等7个已知化合物,以及5’-O-methydioncopeltine、isoquinoline phylline 2个新化合物。 徐智秀等[9]以反相高效液相色谱法分离了9种人参皂苷(I), 利用三级四级杆质谱研究了9种I的一级质谱(主要给出相对分子质量信息)和二级质谱(提供碎片结构信息),通过它们的质谱图差异对其进行了鉴别, 并将方法用于实际样品中的9种I的定性。 郭继芬等[10]选用Discovery C18柱,以甲醇-水-甲酸(40:60:0.025)为流动相,经紫外检测后,在ESI- 扫描方式下,对HPLC—UV图谱中各色谱峰进行一级和二级质谱分析,与对照品比较鉴定了提取物中4个已知的黄酮类化合物,推断出3个未知黄酮苷类化合物可能的结构。 2液相色谱质谱联用在环境分析上的应用 1
液相色谱串联质谱联用仪检测技术 实验指导 (2014、2015级) 课程内容(一个实验8学时): (1)AB Sciex Qtrap 4500 三重四级杆/离子阱液相色谱串联质谱联用仪的结构原理、操作及定性定量应用。 (2)利用液相色谱串联质谱联用仪快速测定水果中7种农药的残留量。 吉林农业大学农业部参茸质检中心 2017.03
实验一AB Sciex Qtrap 4500 三重四级杆/离子阱液相色谱串联质谱联用仪的结构原理、操作及定性定量应用 一.实验目的和意义 通过学习液质联用仪的构成和使用方法,及其在定性、定量分析中的应用,培养学生使用液质联用仪进行仪器分析的能力,并培养学生严谨的科学态度、细致的工作作风、实事求是的数据报告和良好的实验习惯(准备充分、操作规范,记录简明,台面整洁、实验有序,良好的环保和公德意识)。培养培养学生的动手能力、理论联系实际的能力、统筹思维能力、创新能力、独立分析解决实际问题的能力、查阅手册资料并运用其数据资料的能力以及归纳总结的能力等。 (一)检测仪器 1、仪器名称高效液相色谱串联质谱联用仪(简称LC-MS-MS)。型号:4500 QTRAP(美国Applied Biosystems公司)。 2、仪器组成液相色谱部分:岛津LC-30A,配有在线脱气机、超高压二元泵、自动进样器;串联质谱部分:QTRAP4500,配有ESI离子源、串联四级杆/线性离子阱。 3、主要性能指标离子化方式:ESI电离质量范围:(5 ~ 1700)amu 分辨率:> 6900 质量稳定性:0.1 amu/12h 灵敏度:1pg reserpine, ESI+, MRM扫描(m/z : 609/195),信噪比S/N > 120:1 扫描速度:4000 amu/sec 质量准确度:< 0.01%(全质量数范围) 4、方法原理高效液相色谱二元泵将流动相泵人系统并混合,自动进样器将待测样品注入流动相中,随流动相进入色谱柱,由于样品不同组分在色谱柱中保留时间不同,各组分被分开,依次进入离子源。在离子源中,各组分以ESI或APCI方式电离,被加速后进入质量分析器。4500QTRAP 的质量分析器主要由Q1、Q2、Q3三组四级杆串联组成。Q1可将分子离子按质荷比(m/z)大小分开;Q2是碰撞室,可将母离子进一步破碎为碎片离子;Q3具有四级杆和线性离子阱两种功能,作为四级杆时可将分子离子或碎片离子按质荷比大小分开,作为离子阱还可富集离子从而提高检测灵敏度。各组分的不同离子在质量分析器中被破碎、分离,并按质荷比大小依次抵达监测器,经记录即得到按不同质荷比排列的离子质谱图。4500QTRAP通过串联四级杆/线性离子阱两种不同质谱技术的结合,可以在单次分析中对复杂样本中的单个成分同时进行定性和定量,也可以对多个化合物进行定量分析。整台仪器的控制、数据采集、数据处理、结果输出均由PC计算机Windows操作系统支持下的Analyst软件控制完成。
液相色谱-质谱联用技术(LC-MS)的各种模式探索 一、实验目的 1、了解LC-MS的主要构造和基本原理; 2、学习LC-MS的基本操作方法; 3、掌握LC-MS的六种操作模式的特点及应用。 二、实验原理 1、液质基本原理及模式介绍 液相色谱-质谱法(Liquid Chromatography/Mass Spectrometry,LC-MS)将应用范围极广的分离方法——液相色谱法与灵敏、专属、能提供分子量和结构信息的质谱法结合起来,必然成为一种重要的现代分离分析技术。 但是,LC是液相分离技术,而MS是在真空条件下工作的方法,因而难以相互匹配。LC-MS经过了约30年的发展,直至采用了大气压离子化技术(Atmospheric pressure ionization,API)之后,才发展成为可常规应用的重要分离分析方法。现在,在生物、医药、化工、农业和环境等各个领域中均得到了广泛的应用,在组合化学、蛋白质组学和代谢组学的研究工作中,LC-MS 已经成为最重要研究方法之一。 质谱仪作为整套仪器中最重要的部分,其常规分析模式有全扫描模式(Scan)、选择离子监测模式(SIM)。 (一)全扫描模式方式(Scan):最常用的扫描方式之一,扫描的质量范围覆盖被测化合物的分子离子和碎片离子的质量,得到的是化合物的全谱,可以用来进行谱库检索,一般用于未知化合物的定性分析。实例:(Q1 = 100-259m/z) (二)选择离子监测模式(Selective Ion Monitoring,SIM):不是连续扫描某一质量范围,而是跳跃式地扫描某几个选定的质量,得到的不是化合物的全谱。主要用于目标化合物检测和复杂混合物中杂质的定量分析。实例:(Q1 = 259m/z) 本实验采用三重四极杆质谱仪(Q1:质量分析器;Q2:碰撞活化室;Q3:
气相色谱法-质谱联用 气相色谱法–质谱法联用(英语:Gas chromatography–mass spectrometry,简称气质联用,英文缩写GC-MS)是一种结合气相色谱和质谱的特性,在试样中鉴别不同物质的方法。GC-MS的使用包括药物检测(主要用于监督药物的滥用)、火灾调查、环境分析、爆炸调查和未知样品的测定。GC-MS也用于为保障机场安全测定行李和人体中的物质。另外,GC-MS 还可以用于识别物质中以前认为在未被识别前就已经蜕变了的痕量元素。 GC-MS已经被广泛地誉为司法学物质鉴定的金标方法,因为它被用于进行“专一性测试”。所谓“专一性测试”就是能十分肯定地在一个给定的试样中识别出某个物质的实际存在。而非专一性测试则只能指出试样中有哪类物质存在。尽管非专一性测试能够用统计的方法提示该物质具体是那种物质,但存在识别上的正偏差。 目录 1 历史 2 仪器设备 2.1 GC-MS吹扫和捕集 2.2 质谱检测器的类型 3 分析 3.1 MS全程扫描 3.2 选择的离子检测 3.3 离子化类型 3.3.1 电子离子化 3.3.2 化学离子化 3.4 GC-串联MS 4 应用 4.1 环境检测和清洁 4.2 刑事鉴识 4.3 执法方面的应用
4.4 运动反兴奋剂分析 4.5 社会安全 4.6 食品、饮料和香水分析 4.7 天体化学 4.8 医药 5 参考文献 6 参考书目 7 外部链接 历史用质谱仪作为气相色谱的检测器是上个世纪50年代期间由Roland Gohlke和Fred McLafferty首先开发的。当时所使用的敏感的质谱仪体积庞大、容易损坏只能作为固定的实验室装置使用。 价格适中且小型化的电脑的开发为这一仪器使用的简单化提供了帮助,并且,大大地改善了分析样品所花的时间。1964年,美国电子联合公司(Electronic Associates, Inc. 简称EAI)-美国模拟计算机供应商的先驱在开始开发电脑控制的四极杆质谱仪Robert E. Finnigan的指导下[3]开始开发电脑控制的四极杆质谱仪。到了1966年,Finnigan和Mike Uthe的EAI分部合作售出500多台四极杆残留气体分析仪。1967年,Finnigan仪器公司the (Finnigan Instrument Corporation,简称FIC)组建就绪,1968年初就给斯坦福大学和普渡大学发送了第一台GC/MS的最早雏型。FIC最后重新命名为菲尼根公司(Finnigan Corporation)并且继续持世界GC/MS系统研发、生产之牛耳。 1966年,当时最尖端的高速GC-MS (the top-of-the-line high-speed GC-MS units)单元在不到90秒的时间里,完成了火灾助燃物的分析,然而,如果使用第一代GC-MS至少需要16分钟。到2000年使用四极杆技术的电脑化的GC/MS仪器已经化学研究和有机物分析的必不可少的仪器。今天电脑化的GC/MS仪器被广泛地用在水、空气、土壤等的环境检测中;同时也用于农业调控、食品安全、以及医药产品的发现和生产中。 气质联用色谱是由两个主要部分组成:即气相色谱部分和质谱部分。气相色谱使用毛细管柱,其关键参数是柱的尺寸(长度、直径、液膜厚度)以及固定相性质(例如,5%苯基
气相色谱质谱联用原理 和应用 WTD standardization office【WTD 5AB- WTDK 08- WTD 2C】
气相色谱-质谱联用测定农药多残留 摘要:本文研究了气相色谱-质谱联用(GS-MS)仪检测农药残留的方法,辅助以样品前处理技术,对蔬菜、水果、食用油、土壤中的农药多残留的检测方法进行了研究,取得了比较理想的效果。 关键词:气相色谱-质谱联用仪;农药多残留;检测 1引言 当前人类环境持续恶化,世界各国在工业、民用、科技、商业和军事防御等领域都面临着严重的环境污染问题。随着人们对环境污染、食品安全的关注,环境、食品中有机污染物检测方面的规范越来越严格,相应的检测技术也越来越先进。在各种有机物检测技术中,色谱仪器与质谱仪器联用作为一种比较成熟的检测手段,既可发挥色谱法的高分离能力,又兼具质谱准确鉴定化合物结构的优点,即可定性又可定量,尤其适用于环境样品中微量、痕量有机污染物的分析检测工作。1979 年美国环保局(EPA)将GC-MS(Gas Chromatography-Mass Spectrometry)联用技术列为检测饮用水、地表水中有机物的标准分析方法。随着仪器的不断完善与发展,检测技术的成熟与推广,GC-MS 法应用范围越来越广。除了在传统挥发油、脂肪油等的分析测定方面不断发展与普及外,在环境有机污染物检测、食品安全、农药残留、化妆品禁用成分研究等方面的应用也得到了广泛开展。 近年来,由于农药的大量使用引起的食品安全问题已被人们广泛的认识、关注和重视。人们食用了受到农药严重污染的蔬菜水果,而造成人体急性中毒或者慢性中毒的事件屡有发生。为保证食品的质量,世界卫生组织和世界各国制订了严格的限量标准,与此同时,许多国家也借此施行技术壁垒,使得农药残留问题不仅是影响人的身体健康,而且也严重影响到国家的对外贸易。 由于各类食品组成成分复杂,不同农药品种的理化性质存在较大差异,并且近年来高效、低毒、低残留农药品种不断涌现,给农药残留检测技术提出了更高的要求。发展快速、可靠、灵敏和实用的农药残留分析技术无疑是控制农药残留、保证食品安全和避免国际间有关贸易争端的基础。目前,我国农药残留限量标准制定工作滞后,残留监测体系不健全,残留检测能力有限、覆盖面窄。因此,我国应该根据自己的技术条件及农产品市场制定相应的多残留分析方法。 食品中的农药残留污染影响着人民生活质量的提高和食品贸易的顺利进行。日常食用的果蔬施用的农药种类繁多,常见的农药如有机磷类农药、氨基甲酸酯类农药、菊酯类农药和除草剂,抑菌剂等。由于果蔬中往往同时残留不同种类的农药,这对多残留同时检测条件提出很高要求。由于气相色谱-质谱联用( GC-MS) 具有灵敏度
液相色谱-串联质谱(LC/MS/MS)法测定癫痫患者血清中 卡马西平的浓度 谢 华ì,王 荣,贾正平?, 徐丽婷 (兰州军区兰州总医院临床药理基地,兰州 730050) 摘要目的:本文建立了液相色谱-串联质谱(LC/MS/MS)法测定患者血清中的卡马西平浓度的方法。方法:色谱柱:Zorbax Extend-C18柱(150×4.6 mm I.D,5μm);流动相:甲醇-0.01mmol·L-1乙酸 胺溶液(80:20,v/v);流速:0.3 mL·min-1。结果:卡马西平浓度在2~40 ng·mL-1范围内,峰面积与浓度线性关系良好,平均回收率为101.1%,日内精密度、日间精密度的RSD分别为3.39%和4.11%。并测定了10名患者血清中卡马西平的浓度。结论:本方法具有良好的灵敏度、准确度、精确度及专属性,结果准确,重现性好,易于操作,可用于患者血清中卡马西平浓度的测定。 关键词卡马西平;LC/MS/MS;血清 Content Determination of Carbamazepine in epileptic patient serum by Liquid Chromatographic Tandem Mass Spectrometry XIE Hua, JIA Zheng-ping*, WANG Rong, XU Li-ting (Base of Clinic Pharmacology, Lanzhou General Hospital, Lanzhou Command, Lanzhou 730050, China) ABSTRACT OBJECTIVE:An analytical method based on Liquid Chromatography with tandem Mass Spectrometry (LC-MS/MS) detection was developed for the content determination of carbamazepine in epileptic patient serum. METHODS: The method included that the column was Zorbax Extend-C18(150×4.6 mm I.D.,5μm ); mobile phase was methanol-0.01mmol·L-1amine acetic acid (80:20,v/v) at a flow rate of 0.3 mL·min-1. RESULTS: The method was proved to be linear in the range of 2~40ng·mL-1 with a regression confficient of 0.9976. The average recovery rate was 101.1%(n=5). The RSD of average contents of intra-day and inter-day was 3.39% and 4.11% respectively. The carbamazepine concentrations of ten epileptic patient’s serums were detected. CONCLUSION: This method is accurate, precise, sensitive and specific to be used in the content determination of carbamazepine serum. KEY WORDS Carbamazepine; LC/MS/MS; Serum ?基金项目:国家科技部重大项目(2008ZXJ09014-010) ì主管药师。研究方向:临床治疗药物监测。电话:(0931)8994675; E-mial: xiehua-72@https://www.doczj.com/doc/2b5486429.html, ?通讯作者:教授,主任药师,博士。研究方向:临床药学。电话:(0931)8994652。
液相色谱—质谱联用来进行物质分离的实验 一、实验目的 1.了解液相色谱—质谱联用的基本原理; 2.掌握液相色谱—质谱联用时的操作步骤及实验方法; 3.学习分析色谱图和质谱图。 二、实验原理 利用不同的物质在固定相和流动相中具有不同的分配系数,当两相作相对位移时,使这些物质在两相间进行反复多次分配, 使得原来微小的分配差异产生明显的分离效果,从而依先后次 序流出色谱柱,以此来达到分离多种物质的目的。然后依次流 出的物质进入质谱中被打碎成为各种离子而被检测到。以此达 到分离的目的。 三、实验仪器和材料 高效液相色谱仪及质谱仪(见下图)、甲醇、水、TADB(相对分子量516)、TAIW(相对分子量336)、色谱柱
四、实验步骤 1.将待分离的两种物质的混合物配成溶液加入到2号样瓶中去; 2.启动联机软件,在四元泵模块的空白处右键单击,在弹出的 “方法”选项中编辑好流动相和流速,点击确定,以使体系过 渡到目标状态,直到压力稳定为止; 3.进入“方法”菜单,“编辑完整方法菜单”,按照“方法参考”进行编 辑(“方法参考”中的参数编辑完成后继续进行编辑,编辑质 谱的相关参数:选择正负极及电压等),编辑完成后再次进 入“方法菜单”,选择“方法另存为”命名后点击“确定”进入“序列” 菜单,“序列表菜单”,然后编辑样品瓶位置为1号、样品名称、 使用方法、进样次数、数据文件、进样量,确定后再次进入 “序列菜单”的“序列参数”菜单,再选择文件夹,确定; 4.方法编辑完成且压力稳定后,点击进样器左上方的“序列/开 始序列”按钮,进行测试,等待测试完毕,点击停止按钮。 然后进入“脱机”软件,查看积分测试报告。 五、实验结果及分析 实验时的液相色谱条件统一为:70%的甲醇,流速0.4ml/min,进样量1ul,波长230nm,测试时间15min。在正极性条件下: