
C.P.中国药典
(2)溶解度是药品的一种物理性质。各品种项下选用的部分溶剂及其在该溶剂中的溶解性能,可供精制或制备溶液时参考;对在特定溶剂中的溶解性能需作质量控制时,在该品种检查项下另作具体规定。药品的近似溶解度以下列名词术语表示:
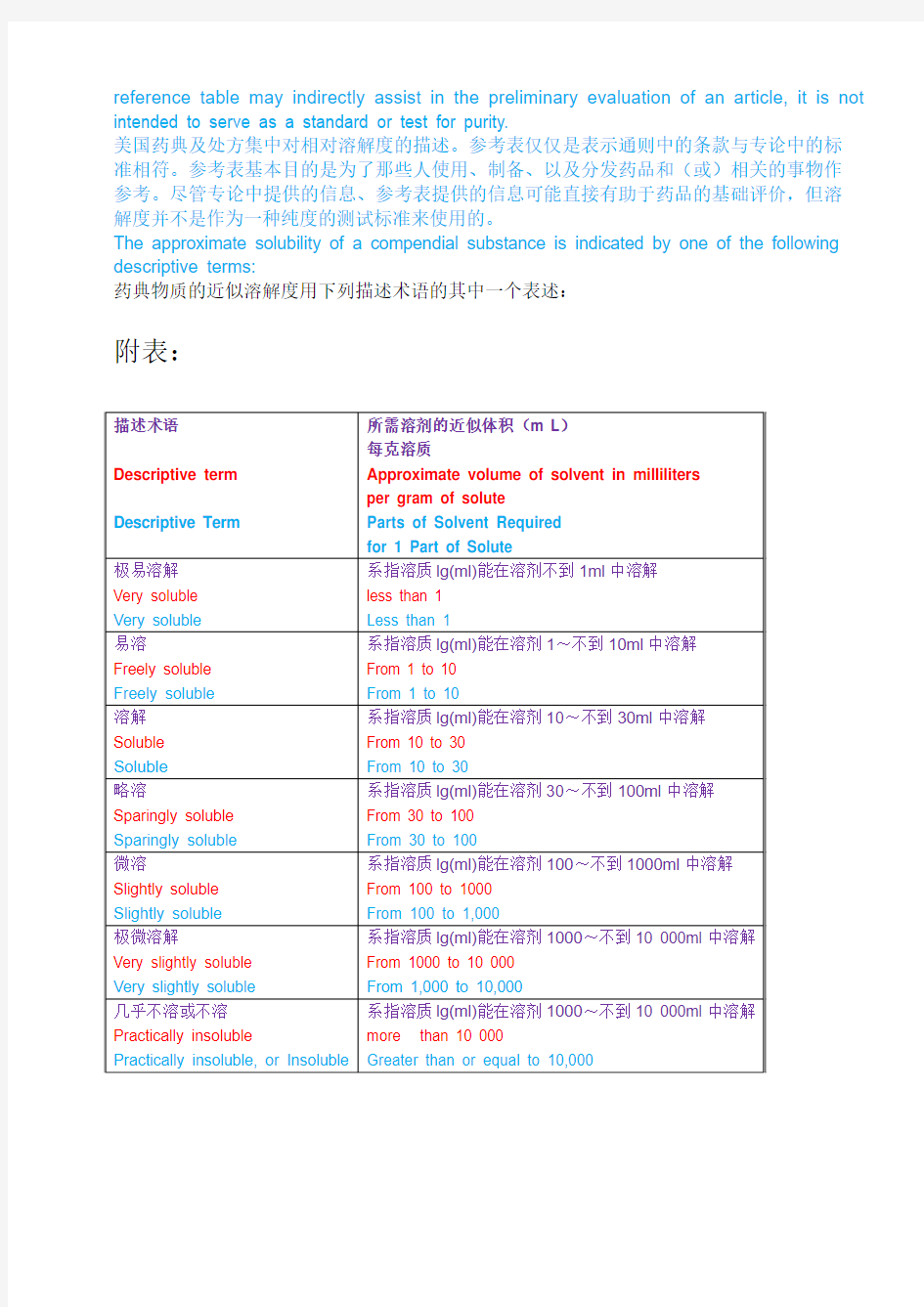
极易溶解:系指溶质lg(ml)能在溶剂不到1ml中溶解;
易溶:系指溶质lg(ml)能在溶剂1~不到10ml中溶解;
溶解:系指溶质lg(ml)能在溶剂10~不到30ml中溶解;
略溶:系指溶质lg(ml)能在溶剂30~不到100ml中溶解;
微溶:系指溶质lg(ml)能在溶剂100~不到1000ml中溶解;
极微溶解:系指溶质lg(ml)能在溶剂1000~不到10 000ml中溶解;
几乎不溶或不溶:系指溶质lg(ml)在溶剂10 000ml中不能完全溶解。
试验法:除另有规定外,称取研成细粉的供试品或量取液体供试品,于25C±2°C—定容量的溶剂中,每隔5分钟强力振摇30秒钟;观察30分钟内的溶解情况,如无目视可见的溶质颗粒或液滴时,即视为完
全溶解。
E.P.欧洲药典
Solubility.溶解性:In statements of solubility in the Characters section, the terms used have the following significance, referred to a temperature between 15 °C and 25 °C. 在性状部分,溶解性的表述中,使用术语所含如下意思,是指温度15 °C 至25 °C.
U S P. 美国药典
5.30. Description and Solubility溶解度描述
Only where a quantitative solubility test is given in a monograph and is designated as such is it a test for purity.只有在专论中给出定量的溶解度检验,才会认为它(溶解度)是一种纯度检查。
A monograph may include information regarding the article’s description. Information about an article’s “description and solubility” also is provided in the reference table
专论可能有关于通则中所描述的信息。通则中“溶解度描述”的信息也可能出现参考表中。Description and Relative Solubility of USP and NF Articles. The reference table merely denotes the properties of articles that comply with monograph standards. The reference table is intended primarily for those who use, prepare, and dispense drugs and/or related articles. Although the information provided in monographs and the information in the
reference table may indirectly assist in the preliminary evaluation of an article, it is not intended to serve as a standard or test for purity.
美国药典及处方集中对相对溶解度的描述。参考表仅仅是表示通则中的条款与专论中的标准相符。参考表基本目的是为了那些人使用、制备、以及分发药品和(或)相关的事物作参考。尽管专论中提供的信息、参考表提供的信息可能直接有助于药品的基础评价,但溶解度并不是作为一种纯度的测试标准来使用的。
The approximate solubility of a compendial substance is indicated by one of the following descriptive terms:
药典物质的近似溶解度用下列描述术语的其中一个表述:
附表:
一、目的: 二、范围: 本标准适用于样品羟值的测定。 三、职责: 1、检验员:严格按操作规程操作,认真、及时、准确地填写检验记录; 2、化验室负责人:监督检查检验员执行本操作规程。 四、内容: 1、定义:羟值系指供试品1g中含有的羟基,经用以下方法酰化后,所需氢氧化钾的重量(mg)。 1.1仪器: 电子天平(万分之一)、具塞锥形瓶(250ml)、胖肚移液管(5ml,A级)、量筒(20ml)、恒温水浴锅、碱式滴定管(50ml,A级)、滴管。 1.2试剂: 1.2.1吡啶AR 1.2.2氢氧化钠滴定液(1mol/L):见EK/SOP-QC8005氢氧化钠滴定液(1、0.5、0.1mol/L)配制与标定操作规程 1.2.3甲酚红-麝香草酚蓝混合指示液:见EK/SOP-QC8003指示剂与指示液配制操作规程1.2.4吡啶-水(3:5):取吡啶30ml和水50ml混合均匀,即得。 1.2.5酰化剂:取对甲苯磺酸14.4g,置500ml碘瓶中,加乙酸乙酯360ml,振摇溶解后,缓缓加入醋酐120ml,摇匀,放置3日后备用。 1.3测定方法: 除另有规定外,按表中规定的重量,精密称取供试品,置250ml的干燥碘瓶中,精密加入酰化剂5ml,用吡啶少许湿润瓶塞,稍拧紧,轻轻摇动使完全溶解,置50℃±1℃水浴中25分钟(每10分钟轻轻摇动)后,放冷,加吡啶-水(3:5)20ml,5分钟后加甲酚红-麝香草酚蓝混合指示液8~10滴,用氢氧化钾(或氢氧化钠)滴定液(1mol/L)滴定至溶液显灰蓝色或蓝色;同时做空白试验。 1.4计算结果: D W F 1. 56 A - B + ? ? = ) ( 供试品的羟值 式中: B为空白试验消耗氢氧化钠滴定液(1mol/L)的体积(ml); A为供试品消耗氢氧化钠滴定液(1mol/L)的体积(ml); 羟值检验操作规程 编写/修订人/日期年月日部门/姓名 审核人/日期年月日部门/姓名 批准人/日期年月日部门/姓名 执行日期2020年11月01日颁发部门品质部 分发部门品质部
2015版《中国药典》考卷 一、填空题(20分) 1、《中华人民共和国药典》(以下简称《中国药典》)2015年版已由国家食品药品监督管理总局2015年第67号公告(2015年07月15日)发布,自起实施。 2、2015版药典将分为四部出版,每部的主要内容分别是一部;二部;三部;四部、。 3、山药等10种传统习用硫磺熏蒸的中药材及其饮片,二氧化硫残留量不得过,其他中药材及其饮片的二氧化硫残留量不得过。 4、“”项下明确列出的有机溶剂或未在正文中列有此项检查的品种,如生产过程中引入或产品中残留有机溶剂,均应按附录“”检查并应符合相应溶剂的限度要求。 5、微生物计数方法:1:;2:;3:最可能数法。 6、常用的鉴别方法包括和。 7、含量测定中常用的方法有和。 8、药品的灰分测定主要是指和。 9、重金属测定主要的测试方法有和。 10、SO2的测定方法有、和离子色谱法。 二、选择题(20分) 1、在《中国药典》检定通则中规定,以下哪种中药材的SO2残留量不得超过400 mg/kg。() A、山药 B、山药片 C、天冬 D、白芍 2、2015版《中国药典》四部通则2331 二氧化硫残留量测定法中规定三种方法,以下哪种不属于规定的方法。() A、酸碱滴定法 B、离子色谱法 C、液相色谱法 D、气相色谱法 3、以下哪种元素不属于重金属元素。() A、铅 B、钙 C、砷 D、磷 4、《中国药典》中通则0832水分测定法中明确了5种方法,除烘干法、减压干燥法外,以下哪种方法不是水分测定的方法。() A、费休氏法 B、甲苯法 C、气相色谱法 D、液相色谱法 5、在《中国药典》中规定除矿物、动物、海洋类以外的中药材中,铜的限值是。() A、10 mg/kg B、5 mg/kg C、1 mg/kg D、20 mg/kg 6、以下哪种测定方法不是《中国药典》规定的方法。() A、水溶浸出物测定法 B、醇溶性浸出物测定法 C、挥发性醚浸出物测定法 D、酯溶性浸出物测定法 7、下面哪种化学物质不是农药。() A、六六六 B、艾氏剂 C、氯丹 D、DNT 8、下面哪种农药不是有机氯类农药。() A、艾氏剂 B、狄氏剂 C、七氯 D、乐果 9、茯苓的SO2限值要求是。() A、150 mg/kg B、400 mg/kg C、10 mg/kg D、100 mg/kg 10、以下哪个选项不是气相色谱仪中的组件。() A、色谱柱 B、流动相 C、氦气 D、进样器 三、判断题(20分) 1、人参对农药残留量只有六六六、滴滴涕、五氯硝基苯有限定要求。()
2020年版《中国药典》通则调整—0121 贴剂 (蓝色字体表示新增内容,红色字体表示删减内容) 贴剂系指原料药物与适宜的材料制成的、供粘贴粘贴贴敷在皮肤上的,可产 生全身性或局部作用的一种薄片状柔性制剂。 贴剂有背衬层、药物贮库、黏贴层及临用前需除去的保护层。贴剂可用于完 整皮肤表面,也可用于有疾患或不完整的皮肤表面。其中用于完整皮肤表面,能将 药物输送透过皮肤进入血液循环系统起全身作用的贴剂称为透皮贴剂。 透皮贴剂通过扩散而起作用,药物从贮库中扩散直接进入皮肤和血液循环,若有控释膜层和黏贴层则通过上述两层进入皮肤和血液循环。透皮贴剂的作用时间 由其药物含量及释药速率所决定。其释放速度受到药物浓度影响。 贴剂的贮库可以是骨架型或控释膜型。 贴剂通常由含有活性物质的支撑层和背衬层以及覆盖在药物释放表面上的 保护层组成;保护层起防粘和保护制剂的作用,通常为防粘纸,塑料或金属材料, 当除去时,应不会引起贮库及黏贴层等的剥离。贴剂的保护层、活性成分不能透过,通常水也不能透过。 根据需要,贴剂可使用药物贮库、控释膜或黏附材料。 当用于干燥、洁净、完整的皮肤表面,用手或手指轻压,贴剂应能牢牢地贴于皮肤表面,从皮肤表面除去时应不对皮肤造成损伤,或引起制剂从背衬层剥离。 贴剂在重复使用后对皮肤应无刺激或引起过敏。 贴剂在生产与贮藏期间应符合下列有关规定。 一、贴剂所用的材料及辅料应符合国家标准有关规定,无毒、无刺激性、性质稳定、与原料药物不起作用。并应考虑到对贴剂局部刺激性和药物性质的影响。 常用的材料为铝箔-聚乙烯复合膜、防粘纸、乙烯-醋酸乙烯共聚物、丙烯酸或聚异丁烯压敏胶、硅橡胶和聚乙二醇等。 二、贴剂根据需要可加入表面活性剂、乳化剂、保湿剂、防腐剂、抗氧剂或 透皮促进剂等。 三、贴剂外观应完整光洁,有均一的应用面积,冲切口应光滑,无锋利的边缘。
总则 一、《中华人民共和国药典》简称《中国药典》,依据《中华人民共和国药品管理法》组织制定和颁布实施。《中国药典》一经颁布实施,其同品种的上版标准或其原国家标准即同时停止使用。 《中国药典》由一部、二部、三部、四部及其增补本组成。一部收载中药,二部收载化学药品,三部收载生物制品,四部收载通则和药用辅料。 本部为《中国药典》四部。 二、国家药品标准由凡例与正文及其引用的通则共同构成。药典收载的凡例与通则对未载入本版药典但经国务院药品监督管理部门颁布的其他中药标准具同等效力。 三、凡例是正确使用《中国药典》进行药品质量检定的基本原则,是对《中国药典》正文、通则及与质量检定有关的共性问题的统一规定。 四、正文中引用的药品系指本版药典收载的品种,其质量应符合相应的规定。 五、正文所设各项规定是针对符合《药品生产质量管理规范》(Good Manufacturing Practices,GMP)的产品而言。任何违反GMP或有未经批准添加物质所生产的药品,即使符合《中国药典》或按照《中国药典》没有检出其添加物质或相关杂质,亦不能认为其符合规定。 六、《中国药典》的英文名称为Pharmacopoeia of The People's Republic of China;英文简称为Chinese Pharmacopoeia;英文缩写为ChP。 七、《中国药典》各品种项下收载的内容统称为标准正文,正文系根据药物自身的理化与生物学特性,按照批准的来源、处方、制法和贮藏、运输等条件所制定的、用以检测药品质量是否达到用药要求并衡量其质量是否稳定均一的技术规定。 正文 八、《中国药典》各品种项下收载的内容统称为标准正文,正文系根据药物自身的理化与生物学特性,按照批准的来源、处方、制法和贮藏、运输等条件所制定的、用以检测药品质量是否达到用药要求并衡量其质量是否稳定均一的技术规定。 九、药用辅料标准正文内容一般包括:(1)品名(包括中文名、汉语拼音与英文名);(2)有机物的结构式; (3)分子式、分子量与CAS编号;(4)来源;(5)制法;(6)性状;(7)鉴别;(8)理化检查;(9)含量测定;(10)类别;(11)贮藏;(12)标示等。 通则 十、通则主要收载制剂通则、通用检测方法和指导原则。制剂通则系按照药物剂型分类,针对剂型特点所规定的基本技术要求;通用检测方法系各正文品种进行相同检查项目的检测时所应采用的统一的设备、程序、方法及限度等;指导原则系为执行药典、考察药品质量、起草与复核药品标准等所制定的指导性规定。 名称及编排 十一、正文收载的药品中文名称通常按照《中国药品通用名称》收载的名称及其命名原则命名,《中国药典》收载的药品中文名称均为法定名称;本版药典收载的原料药英文名除另有规定外,均采用国际非专利药名(International Nonproprietary Names,INN)。 有机药物的化学名称系根据中国化学会编撰的《有机化学命名原则》命名,母体的选定与国际纯粹与应用化学联合会(International Union of Pure and Applied Chemistry,IUPAC)的命名系统一致。 十二、药品化学结构式按照世界卫生组织(World Health Organization,WHO)推荐的“药品化学结构式书写指南”书写。 十三、正文按药品中文名称笔画顺序排列,同笔画数的字按起笔笔形一丨丿丶乛的顺序排列;通则包括制剂通则、通用检测方法和指导原则,按分类编码;索引分按汉语拼音顺序排序的中文索引以及英文名和中文名
精心整理 制药企业产品检测理论试题 一、单选题 1下列哪项不属于2015版《中国药典》一部正文收载内容?(C ) 2 A.药材和饮片 B.成方制剂和单味制剂 C.药用辅料 D.提取物 E.植 3 4 A. 5 6 A. 7 C. 8 9 A. 10 11 12)13 A. 14下列有关【贮藏】项下的规定,描述错误的是(D ) 15 A.冷处是指2~10℃ B.常温系指10~30℃ 16 C.阴凉处系指不超过10℃ D.密闭的目的是防止风化、吸潮、挥发或异 物进入 17试验中供试品与试药等“称重”或“量取”的量,均以阿拉伯数字表示,其精确度可根据述职的有效数位来确定,下列描述错误的是(A )
精心整理 18 A.如称取“0.1g”系指称取重量可为0.05~0.16g; 19 B.称取“2g”,系指称取重量可为1.5~2.5g; 20 C. 称取“2.0g”,系指称取重量可为1.95~2.05g; 21 D.称取“2.00g”,系指称取重量可为1.995~2.005g。 222015版《中国药典》规定,细粉系指能全部通过五号筛,并含能通过六号筛不少于的粉末。(D ) 23 24 25 A. 26 27 A. 28 29 A. 30 31 32 33 A. 34 C.水的纯度、是否含含有离子杂质、pH和温度 D.水是否含有离子杂质、pH和 温度 352015版《中国药典》可见异物检查法中,5瓶注射用无菌冻干粉制剂如检出微细可见异物,每瓶中检出微细可见异物数量不得过( C )。 36 A.1个 B.2个 C.3个 D.4个 E.5个 37原料药与制剂稳定性试验考察中加速试验一般要求的温湿度为(A ) 38 A.40℃±2℃;75%±5%; B. 25℃±2℃;60%±5%;
一、目的: 制订详尽的工作程序,规范检验操作,保证检验数据的准确性。 二、范围: 本标准适用于参考美国药典标准检验品种重金属的测定。 三、职责: 1、检验员:严格按操作规程操作,认真、及时、准确地填写检验记录; 2、化验室负责人:监督检查检验员执行本操作规程。 四、内容: 1、特殊试剂: 1.1硝酸铅原液:将159.8毫克的硝酸铅溶于100毫升水中,加入1毫升硝酸,然后用水稀释至1000毫升。制备此溶液并将其储存在无可溶性铅盐的玻璃容器中。 1.2标准铅溶液:临用新制,用水稀释10.0毫升硝酸铅原液至100.0毫升。每毫升标准铅溶液含有相当于10微克的铅。以每克被测物质100微升标准铅溶液为基础制备的对比溶液包含相当于每百万份被测物质1部分的铅。 2、方法一: 2.1 pH 3.5乙酸盐缓冲液:溶解25克醋酸铵在25毫升水中,加入6mol/l盐酸38毫升。如果需要调节,可用6mol/l氢氧化铵或6mol/l盐酸调节pH值为3.5,用水稀释至100毫升,并混合。 2.2标准制备:将标准铅溶液(20微克铅)2毫升放入50毫升比色管中,用水稀释至25毫升。使用pH计或短程pH指示纸作为外部指示剂,用1mol/l乙酸或6mol/l氢氧化铵调节到 3.0到 4.0之间的pH,用水稀释至40毫升,混匀。 2.3供试品制备:按照各专著的指示,将试验准备的溶液放入50mL比色管中,或使用各专著中指定体积的酸,溶于水中,用水稀释至25mL,单位为按公式计算的待测物质: 2.0/(1000L) 其中L是重金属限度,占百分数。使用pH计或短程pH指示剂纸作为外部指示剂,用1mol/l 乙酸或6 mol/l氢氧化铵调节pH值在3-4之间,用水稀释至40毫升,并混合。 2.4 监测制备:在第三根50mL比色管中,放入按供试品制备指示制备的溶液25mL,并加入2.0mL标准铅溶液。使用pH计或短程pH指示剂纸作为外部指示剂,用1mol/l乙酸或6mol/l氢氧化铵调节pH值在3-4之间,用水稀释至40毫升,并混合。 2.5方法:在含有标准制剂、供试品制剂和监测制剂的三个试管中,加入2毫升pH 3.5的乙酸缓冲液,然后加入1.2毫升硫代乙酰胺-甘油基TS,用水稀释至50毫升,混合,静置2分钟,在白色表面向下观察:来自试验制剂的溶液的颜色不比来自标准制剂的溶液的颜色深,来自监测制剂的溶液的颜色等于或比来自标准制剂的溶液的颜色深。[注--如果监视器制剂的颜色比标准制剂的颜色浅,则对被测试物质使用方法II而不是方法I]。 3、方法二: 3.1注:此方法不回收汞。
水分测定方法有许多种,我们在选择时要根据食品的性质来选择。常采用的水份测定方法如下: 1、热干燥法:①常压干燥法(此法用的广泛); ②真空干燥法(有的样品加热分解时用); ③红外线干燥法; ④真空器干燥法(干燥剂法); 2、蒸馏法 3、卡尔费休法 4、水分活度AW的测定 下面我们分别讲述测定水分的方法。 一、常压干燥法 1、特点与原理 ⑴特点:此法应用最广泛,操作以及设备都简单,而且有相当高的精确度。 ⑵原理:食品中水分一般指在大气压下,100℃左右加热所失去的物质。但实际上在此温度下所失去的是挥发性物质的总量,而不完全是水。 2、干燥法必须符合下列条件(对食品而言): ⑴水分是唯一挥发成分 这就是说在加热时只有水分挥发。例如,样品中含酒精、香精油、芳香脂都不能用干燥法,这些都有挥发成分。 ⑵水分挥发要完全 对于一些糖和果胶、明胶所形成冻胶中的结合水。它们结合的很牢固,不宜排除,有时样品被烘焦以后,样品中结合水都不能除掉。因此,采用常压干燥的水分,并不是食品中总的水分含量。 ⑶食品中其它成分由于受热而引起的化学变化可以忽略不计。 例:还原糖+氨基化合物△→ 变色(美拉德反应)+H2O↑ 还有 H2C4H4O6(酒石酸)+ 2NaHCO3 → NaC4H4O6(酒石酸钠)+2H2O+2CO2
发酵糖(NaHCO3+KHC4H4O6)△→H2O+CO2+ NaKC4H4O6 高糖高脂肪食品不适应 只看符合上面三点就可采用烘箱干燥法。烘箱干燥法一般是在100~105℃下进行干燥。 我们讲的上面三点,应该是具体的具体分析,对于一个分析工作人员,或者是一个技术员,虽然干燥法必须符合三点要求,那么我们在只有烘箱的情况下,而且蓑红样品不见得符合以上讲的三点,难道就不测水分吗? 例如,啤酒厂要经常测啤酒花的水分,啤酒花中含有一部分易挥发的芳香油。这一点不符合我们的第一点要求,如果用烘箱法烘,挥发物与水分同时失去,造成分析误差。此外,啤酒花中的α—酸在烘干过程中,部分发生氧化等化学反应,这又造成分析上的误差,但是一般工厂还是用烘干法测定,他们一般采取低温长时间(80~85℃烘4小时),或者高温短时(105℃烘1小时) 所以应根据我们所在的环境和条件选择合适的操作条件,当然我们应该首先明白有没有挥发物和化学反应等所造成的误差。 3、烘箱干燥法的测定要点 ⑴取样(称样) 在采样时要特别注意防止水分的变化,对有些食品例如奶粉、咖啡等很容易吸水,在称量时要迅速,否则越称越重。 ⑵干燥条件的选择 三个因素:①温度;②压力(常压、真空)干燥;③时间。 一般是温度对热不稳定的食品可采用70~105℃;温度对热稳定的食品采用120~135℃。 4、操作方法 清洗称量皿→烘至恒重→称取样品→放入调好温度的烘箱(100~105℃)→烘1.5小时→于干燥器冷却→称重→ 再烘0.5小时→称至恒重(两次重量差不超过0.002g即为恒重) *油脂或高脂肪样品,由于脂肪氧化,而后面一次重量反而增加,应以前一次重量计算。 *对于易焦化和容易分解的食品,可以选用比较低的温度或缩短干燥时间。
0100 本制剂通则中原料药物系指用于制剂制备的活性物质,包括中药、化学药、生物制品原料药物。中药原料药物系指 饮片、植物油脂、提取物、有效成分或有效部位》化学药原料药物系指化学合成、或来源于天然物质或采用生物技术获 得的有效成分(即原料药);生物制品原料药物系指生物制品原液或将生物制品原液干燥后制成的原粉。 本制剂通则中各剂型、亚剂型并不适用于所有原料药物,而应取决于原料药物特性、临床给药需求以及药品的安 全性、有效性和稳定性等。 本制剂通则适用于中药、化学药和治疗用生物制品(包 括血液制品、免疫血清、细胞因子、单克隆抗体、免疫调节 剂、微生态制剂等)。预防类生物制品,应符合本版药典三部相应品种项下的有关要求。 除另有规定外,生物制品应于2?8X:避光贮存和运输 。 片剂系指原料药物或与适宜的辅料制成的圆形或异形的 片状固体制剂。 中药还有浸膏片、半浸膏片和全粉片等。 片剂以口服普通片为主,另有含片、舌下片、口腔貼 片、咀嚼片、分散片、可溶片、泡腾片、阴道片、阴道泡腾 片、缓释片、控释片、肠溶片与口崩片等。 含片系指含于口腔中缓慢溶化产生局部或全身作用的片剂。 含片中的原料药物一般是易溶性的,主要起局部消炎、杀菌、收敛、止痛或局部麻醉等作用。 舌下片系指置于舌下能迅速溶化,药物经舌下黏膜吸 收发挥全身作用的片剂。 舌下片中的原料药物应易于直接吸收,主要适用于急症 的治疗。 口腔貼片系指粘贴于口腔,经黏膜吸收后起局部或全身作用的片剂。 口腔貼片应进行溶出度或释放度(通则0931)检查。 咀嚼片系指于口腔中咀嚼后吞服的片剂。 咀嚼片一般应选择甘露醇、山梨醉、蔗糖等水溶性辅料作填充剂和黏合剂。咀嚼片的硬度应适宜。 分散片系指在水中能迅速崩解并均勻分散的片剂。 分散片中的原料药物应是难溶性的。分散片可加水分散 后口服,也可将分散片含于口中吮服或吞服。 分散片应进行溶出度(通则0931)和分散均匀性检查。 可溶片系指临用前能溶解于水的非包衣片或薄膜包衣片剂。 可溶片应溶解于水中,溶液可呈轻微乳光。可供口服、外用、含漱等用。 泡腾片系指含有碳酸氢钠和有机酸,遇水可产生气体 而呈泡腾状的片剂。 泡腾片中的原料药物应是易溶性的,加水产生气泡后应能溶解。有机酸一般用枸橼酸、酒石酸、富马酸等。 阴道片与阴遒泡腾片系指置于阴道内使用的片剂。阴道片和阴道泡腾片的形状应易置于阴道内,可借助器具将阴道片送人阴道。阴道片在阴道内应易溶化、溶散或融化、崩解并释放药物,主要起局部消炎杀菌作用,也可给予性激素类药物。具有局部刺激性的药物,不得制成阴道片《 阴道片应进行融变时限检查(通则0922)。阴道泡腾片还应进行发泡量检査。 缓释片系指在规定的释放介质中缓慢地非恒速释放药物的片剂。缓释片应符合缓释制剂的有关要求(通则9013) 并应进行释放度(通则0931)检查。 控释片系指在规定的释放介质中缓慢地恒速释放药物的片剂。控释片应符合控释制剂的有关要求(通则9013)并 应进行释放度(通则0931)检查。 K溶片系指用肠溶性包衣材料进行包衣的片剂。 为防止原料药物在胃内分解失效、对胃的刺激或控制原 料药物在肠道内定位释放,可对片剂包肠溶衣;为治疗结肠 部位疾病等,可对片剂包结肠定位肠溶衣。 肠溶片除另有规定外,应进行释放度(通则0931)检查。 P崩片系指在口腔内不需要用水即能迅速崩解或溶解的片剂。 —般适合于小剂量原料药物,常用于吞咽困难或不配合 服药的患者。可采用直接压片和冷冻干燥法制备。 口崩片应在口腔内迅速崩解或溶解、口感良好、容易吞 咽,对口腔黏膜无刺激性。 除冷冻干燥法制备的口崩片外,口崩片应进行崩解时限检査(通则0921)。对于难溶性原料药物制成的口崩片,还应进行溶出度检査(通则0931)。对于经肠溶材料包衣的颗粒制成的口崩片,还应进行释放度检査(通则 0931)。 采用冷冻干燥法制备的口崩片可不进行脆碎度检査 。
—、范围:本标准规定了微生物限度的检查方法和操作要求;适用于检品 需氧菌总数、霉菌和酵母菌总数、控制菌的检查。 二、 引用标准:《中国药典》( 通则1105-1106) 三、 目录1.微生物限度标准 2.设备.仪器及用具 3?消毒液、稀释剂.试液及培养基 4. 检查总则(通则1105:非无菌产品微生物限度检查:微生 物计数法,通则1106非无菌产品微生物限度检查:控制菌检查法) 5. 微生物计数法检查 6. 控制菌检查法 7. 实验技术 &附件 1. 微生物限度标准 非无菌药用原料及辅料的微生物限度标准 *未做统一规定。 L1成品微生物限度标准
(1).” 一”为不得检出。 (2).目测霉变者以不合格论。 (3).”无”为标准依据或无相应规定。 1.2工艺用水微生物限度标准 1.3内包装材料微生物限度标准 说明:1?”一”为每100 cm2中不得检出。2.目测霉变者以不合格论。3.”无”为标准依据或无相应规定。 2.设施、仪器及用具
2.1、设施: 2丄1?微生物限度检查室及相关设施:微生物计数试验环境应符合微生物限度检查的要求。检验全过程必须严格遵守无菌操作,防止再污染,防止污染的措施不得影响供试品中微生物的检出。单向流空气区域、工作台直及环境应定期进行监测。 2.12其它设备:高压蒸汽灭菌器;细菌培养箱(30?35?);霉菌培养箱(25-280 ;电炉(或其它适宜的加热装置);恒温水浴;电热干燥箱(250~300匕);电冰箱。生化试剂储存箱。 2.2仪器及器mi 2.2.1.菌落计数器;显微镜(1500X);电子天平或药物天平(感量O.lg); pH系列比色计。 222?玻璃器皿:锥形瓶(250?300ml,内装玻璃珠若干).研钵(玻璃或陶瓷制,f 10?12cm)、培养皿(f 9cm).量筒(100ml).试管(18x 180mm)及塞、吸管(lml分度0.01, 10ml分度0.1)、载玻片、盖玻片、玻璃消毒缸(带盖)。 2.2.3新购的玻璃器皿的清洁:先用流水冲洗,浸泡于1%?2%盐酸(工业用)液中约2?6小时,除去游离碱质,再用流水冲洗。用于化学分析的玻璃仪器,需用重洛酸钾清洁液浸泡数分钟后,再用流水冲洗,最后以纯化水涮洗2?3次,晾干备用。 2.3用过的玻璃器皿: 231未被病原微生物污染的器皿:可随时洗涤。用清水冲洗(或浸泡),除容量仪器外, 可用毛刷和肥皂粉,内外刷洗,再用清水涮洗干净,晾干备 用。容量仪器宜用清洁液浸泡或涮洗,再用流水冲洗,最后以纯化水涮洗 2~3 次。
0832
水分测定法1
第一法(费休氏法) A.容量滴定法 本法是根据碘和二氧化硫在吡啶和甲醇溶液中与水定量反应的原理来测定水分。 所用仪 器应干燥,并能避免空气中水分的侵入;测定应在干燥处进行。 费休氏试液的制备与标定 (1)制备 称取碘(置硫酸干燥器内 48 小时以上)110g,置干燥的具塞锥形瓶(或烧瓶) 中,加无水吡啶 l60ml,注意冷却,振摇至碘全部溶解,加无水甲醇 300ml,称定重量,将 锥形瓶(或烧瓶)置冰浴中冷却,在避免空气中水分侵入的条件下,通入干燥的二氧化硫至 重量增加 72g,再加无水甲醇使成 1000ml,密塞,摇匀,在暗处放置 24 小时。 也可以使用市售费休氏试液。市售的费休氏试液可以是无吡啶试剂,或无甲醇试剂;也 可以是由两种溶液临用前混合而成的费休氏试液。 本试液应遮光,密封,阴凉干燥处保存。临用前应标定滴定度。 (2)标定 精密称取纯化水 10~30mg,用水分测定仪直接标定;或精密称取纯化水 l0~ 30mg,置干燥的具塞锥形瓶中,除另有规定外,加无水甲醇 2~5 ml,在避免空气中水分侵 入的条件下,用费休氏试液滴定至溶液由浅黄色变为红棕色,或用电化学方法[如永停滴定 法(通则 0701)等]指示终点;另做空白试验,按下式计算:
F=
式中
W A? B
F 为每 lml 费休氏试液相当于水的重量,mg; w 为称取纯化水的重量,mg; A 为滴定所消耗费休氏试液的容积,ml; B 为空白所消耗费休氏试液的容积,ml。 测定法 精密称取供试品适量(约消耗费休氏试液 1~5 ml) ,除另有规定外,溶剂为 无水甲醇,用水分测定仪直接测定。或精密称取供试品适量,置干燥的具塞锥形瓶中,加溶 剂 2~5 ml,在不断振摇(或搅拌)下用费休氏试液滴定至溶液由浅黄色变为红棕色,或用 永停滴定法(通则 0701)指示终点;另做空白试验,按下式计算: 供试品中水分含量(%)= 式中
( A ? B) F × 100% W
A 为供试品所消耗费休氏试液的容积,ml; B 为空白所消耗费休氏试液的容积,ml; F 为每 lml 费休氏试液相当于水的重量,mg; W 为供试品的重量,mg。 如供试品吸湿性较强,可称取供试品适量置干燥的容器中,密封(可在干燥的隔离箱中 操作) ,精密称定,用干燥的注射器注入适量无水甲醇或其他适宜溶剂,精密称定总重量, 振摇使供试品溶解,测定水分。洗净并烘干容器,精密称定其重量。同时测定溶剂的水分。 按下式计算: 供试品中水分含量(%)= 式中 W1 W2 W3
(W1 ? W3 )C1 ? (W1 ? W2 )C 2 × 100% W2 ? W3
为供试品、溶剂和容器的重量,g; 为供试品、容器的重量,g; 为容器的重量,g;
1
1 每一部药典均由凡例、正文和附录组成。“凡例”是解释和正确地使用药典进行质量检定的基本原则,并把正文品种、附录及质量检定有关的共性问题加以规定。“凡例”中的有关规定具有法定约束力。 2 乙醇在未指明浓度时,均系指95%(ml/ml)的乙醇。 3 杂质的来源途径生产过程引入和储藏过程中引入。 4 《中国药典》2005年版二部片剂的常规检查项目有重量差异、溶出度(释放度)、崩解时限、含量均匀度。 5 注射液的常规检查项目有装量(装量差异)、可见异物、热原或细菌内毒素检查法、无菌、不溶性微粒等。 6 羚羊角顶端部分光滑,内有细孔道直通角尖,习称通天眼。 7 取用量为“约”若干时,系指取用量不得超过规定量的±10% 8 溶化性检查法中,热水温度应按照《中国药典》凡例中规定为70~80℃。 9 《中国药典》2005年版一部性状项下记载品种的外观、质地、横断面、臭、味、溶解度以及物理常数等。 10 中药制剂分析常用的提取方法有:萃取、冷浸、回流、索氏提取、水蒸气蒸溜法、超声提取法。 11 为保证药品检验工作的科学性和规范化,检验原始记录必须用蓝黑墨水或碳素笔书写,做到记录原始、数据真实、字迹清晰、资料完整。 12 检验者接受检品后,首先对检验卡与样品中的品名、批号、生产厂家、检验依据、检验项目、包装、数量、编号等进行核对,确认无误后,按照质量标准及其方法和有关SOP进行检验,并按要求记录。 13 对药物的质量要求,主要考察药品的安全性和有效性。 14 药品质量标准有关质量控制部分主要包括性状、鉴别、检查、含量测定四大项目。 15 列入国家药品标准的药品名称为药品通用名称,该名称不得作为药品商标使用。 16 国家对麻醉药品、精神药品、医疗用毒性药品、放射性药品,实行特殊管理。 17 医疗机构的药剂人员在调配处方,必须经过核对,对处方所列药品不得擅自更改或者代用。对有配伍禁忌或者超剂量的处方,应当拒绝调配;必要时,经处方医师更正或者重新签字,方可调配。 18 药品生产企业必须对其生产的药品进行质量检验;不符合国家药品标准或者不按照省、自治区、直辖市人民政府药品监督管理部门制定的中药饮片炮制规范炮制的,不得出厂。 19 药品监督管理部门设置或者确定的药品检验机构,承担依法实施药品审批和药品质量监督检查所需的药品检验工作。 20 当事人对药品检验机构的检验结果有异议,可以自收到药品检验结果之日起七日内向原药品检验机构或者上一级药品监督管理部门设置或者确定的药品检验机构申请复验,也可以直接向国务院设置或者确定的药品检验机构申请复验。 21 药品检验机构出具虚假检验报告,构成犯罪的,依法追究刑事责任;不构成犯罪的责令改正、给予警告,对单位并处三万元以上五万元以下的罚款;对直接负责的主管人员和其他直接责任人员依法给予降级、撤职、开除的处分,并处三万元以下的罚款;有违法所得的,没收违法所得;情节严重的,撤销其检验资格。 22 地方药品检验机构的设置规划由省、自治区、直辖市人民政府药品监督管理部门提出,报省、自治区、直辖市人民政府批准。 23 药品抽样必须由两名以上药品检查人员实施,并按照国务院规定药品监督管理部门的规定进行抽样;被抽检方应当提供抽检样品,不得拒绝。 24 评价抽验的工作可由药品检验机构承担;监督抽验的抽样工作由药品监督管理部门承担。 25 药品抽样应当在被抽样单位存放药品的现场进行,被抽样单位应当派专人协助抽样。抽样地点由抽样人员根据被抽样单位的特点确定,一般为药品生产企业的成品仓库和药用原、辅料仓库,药品经营企业的仓库和药品零售企业的营业场所,药品使用单位的仓库和药品零售企业的营业场所,以及其他认为需要抽样的场所。 26 抽样人员完成药品抽样后,应当及时将所抽取的样品移交药品检验机构药品检验机构应在核对被抽取样品与所记录的内容相符、承担检验任务的、“药品抽样记录及凭证”、“药品封签”完整等情况下予以收验。 27 药品检验机构接到样品,在取得检验必须的材料后应当按照在法定质量标准、规定周期内完成检验,并出具药品检验报告书。特殊情况需要延期的,应当报告同级药品监督管理部门批准。 28 抽查检验的样品必须按规定留样备用。 29 我国法定的国家药品标准有中国药典和局颁或部颁标准两类。 30 药品监督管理部门设置或确立的药品检验机构,承担依法实施药品审批和药品质量监督检查所需的药品检验工作。 31 "药品检验报告书的表头栏目报告日期应填写。( D ) A. 检验完成的日期 B. 业务管理室主任审签的日期 C. 报告寄出的日期 D. 授权签字人审定签发报告书的日期" 32 "抽查检验分为两种:。( A ) A. 评价抽验和监督抽验 B. 监督抽验和执法抽验 C. 评价抽检和执法检验 D. 监督抽检与例行抽检" 33 "被抽样单位或药品生产企业对药品检验机构的检验结果有异议的,可以自收到药品检验结果之日起个工作日内提出复验申请;逾期申请复验的,药品检验机构将不再受理。( C ) A. 3 B. 5 C. 7 D. 10" 34 "在国内生产并销售的药品必须符合。( A ) A. 国家药品标准 B. 国际药品标准 C. USP D. 行业标准" 35 "检查化学药品胶囊的装量差异时,一般取样量为。( B ) A. 10粒 B. 20粒 C. 30粒 D. 5粒" 36 "药品检验报告书中所填写的药品名称为:。( B )
附录ⅧM 水分测定法 第一法(费休氏法) A.容量滴定法 本法是根据碘和二氧化硫在吡啶和甲醇溶液中能与水起定量反应的原理以测定水分。所用仪器应干燥,并能避免空气中水分的侵入;测定操作宜在干燥处进行。 费休氏试液的制备与标定 (1)制备称取碘(置硫酸干燥器内48 小时以上)110g,置干燥的具塞锥形瓶中,加无水吡啶160ml,注意冷却,振摇至碘全部溶解后,加无水甲醇300ml,称定重量,将锥形瓶置冰浴中冷却,在避免空气中水分侵入的条件下,通入干燥的二氧化硫至重量增加72g,再加无水甲醇使成1000ml,密塞,摇匀,在暗处放置24 小时。 也可以使用稳定的市售卡尔-费休氏试液。市售的试液可以是不含吡啶的其他碱化剂,不含甲醇的其他醇类等;也可以是单一的溶液或由两种溶液混合而成。 本液应遮光,密封,置阴凉干燥处保存。临用前应标定浓度。 (2)标定精密称取纯化水10~30mg,用水分测定仪直接标定。 或精密称取纯化水10~30mg(视费休氏试液滴定度和滴定管体积而定),置干燥的具塞玻瓶中,除另有规定外,加无水甲醇适量,在避免空气中水分侵入的条件下,用本液滴定至溶液由浅黄色变为红棕色,或用电化学方法[如永停滴定法(附录Ⅶ A)等]指示终点;另作空白试验,按下式计算: F=W/(A-B) 式中 F 为每1ml 费休氏试液相当于水的重量,mg; W 为称取重蒸馏水的重量,mg; A 为滴定所消耗费休氏试液的体积,ml; B 为空白所消耗费休氏试液的体积,ml。 测定法精密称取供试品适量,除另有规定外,溶剂为无水甲醇,用水分测定仪直接测定。 或精密称取供试品适量(约消耗费休氏试液1~5ml),置干燥的具塞玻瓶中,加溶剂适量,在不断振摇(或搅拌)下用费休氏试液滴定至溶液由浅黄色变为红
片剂 片剂系指原料药物与适宜的辅料制成的圆形或异形的片状固体制剂。中药还有浸膏片、半浸膏片和全粉片等。片剂以口服普通片(也包括糖衣片、薄膜衣片)为主,另有含片、舌下片、口腔贴片、咀嚼片、分散片、可溶片、泡腾片、阴道片、阴道泡腾片、缓释片、控释片与肠溶片(包括肠溶衣片和结肠定位肠溶衣片)与口崩片等。 对片剂的质量要求除外观应完整光洁、色泽均匀,有适宜的硬度和耐磨性,以及药典品种项下规定的检验项目外,还应检查“重量差异”和“崩解时限”。此外,阴道片应检查“融变时限”,阴道泡腾片应检查“发泡量”,分散片应检查“分散均匀性”,口腔贴片、阴道片、阴道泡腾片和外用可溶片等局部用片剂应检查“微生物限度”。 “重量差异”检查法 1 简述 1.1 本法适用于片剂的重量差异检查。凡规定检查含量均匀度的片剂,不再进行重量差异的检查。 1.2 在片剂生产中,由于颗粒的均匀度和流动性,以及工艺、设备和管理等原因,都会引起片剂重量差异。本项检查的目的在于控制各片重量的一致性,保证用药剂量的准确。 2 仪器与用具 2.1 分析天平感量0.1mg(适用于平均片重0.30g以下的片剂)或感量lmg(适用于平均片重0.30g或0.30g以上的片剂)。 2.2 扁形称量瓶。 2.3 弯头或平头手术镊。 3 操作方法 3.1 取空称量瓶,精密称定重量;再取供试品20片,置此称量瓶中,精密称定。两 ,得平均片重()。20次称量值之差即为20片供试品的总重量,除以 3.2 从已称定总重量的20片供试品中,依次用镊子取出1片,分别精密称定重量,1 / 7 得各片重量。 4 注意事项在称量前后,均应仔细查对药片数。称量过程中, 应避免用手直接接触供试4.1 品。已取出的药片,不得再放回供试品原包装容器内。4.2 遇有检出超出重量差异限度的药片,宜另器保存,供必要时的复核用。糖衣片应在包衣前检查片
水分检测方法 1.目的 使检验人员能够正确对样品规格中需要水分检测的样品进行水分检测。 2.范围 适用于样品规格中需要水分检验的样品。 3.参考文件 3.1 《中国药典2010年版2部》 3.2 GB_50093-2010食品中水分的测定 4.定义 4.1 恒重:取洁净铝制或玻璃制的扁形称量瓶,置于101℃~105℃干燥箱中,瓶盖斜 支于瓶边,加热1h,取出盖好,置干燥器内冷却0.5h称量,并重复干燥至前后 两次质量差不超过2mg,即为恒重。 5.职责 QC负责按照本方法执行对样品的检测 6.程序 6.1 费休氏法 卡尔·费休水分测定法又分为库仑法和容量法。库仑法测定的碘是通过化学反应 产生的,只要电解液中存在水,所产生的碘就会和水以1:1的关系按照化学反应 式进行反应。当所有的水都参与了化学反应,过量的碘就会在电极的阳极区域形 成,反应终止。容量法测定的碘是作为滴定剂加入的,滴定剂中碘的浓度是已知 的,根据消耗滴定剂的体积,计算消耗碘的量,从而计量出被测物质水的含量。 6.1.1 试剂与材料 试剂:碘、无水吡啶、无水甲醇或者市售卡尔-费休氏试液。 6.1.2 仪器和设备 卡尔-费休水分仪、分析天平:感量为0.1mg 6.1.3 容量法 1)费休氏试液的制备与标定 a)制备:称取碘(置硫酸干燥器内48小时以上)110g,置干燥的具塞 锥形瓶中,加无水吡啶160ml,注意摇允至碘全部溶解后,加无水甲 醇300ml。称定重量,将锥形瓶至冰浴中冷却,在避免空气进入的情 况下,通入干燥的二氧化硫至重量增加72g,再加甲醇使成1000ml, 密塞,摇允,在暗处放置24小时。(也可以使用稳定的市售卡尔- 费休氏试液。市售的试液可以是不含吡啶的其他碱化剂,不含甲醇的 其他醇类等;也可以是单一的溶液或者由两种溶液混合而成。)试液 应遮光,密封,置阴凉干燥处保存。临用前应标定浓度。 b)标定:精密称取10~30mg费休氏试液用卡费休仪器直接标定。或取 干燥的具塞玻瓶,精密称入蒸馏水约30mg,除另有规定外加无水甲 醇2~5ml,在避免空气侵入的条件下,用本液滴定至溶液由浅黄色变 为红棕色,或用永停滴定法指示终点;另作空白试验,标定应取3 份
中国药典与国家药品标准的主要内容 一.单选题 1 问题: 测定旋光度时的测定温度为 答案选项A 、15℃ B 、20℃ C 、25℃ D 、30℃ E 、35℃ 标准答案:B 2 问题: 药品贮藏条件的凉暗处是指 答案选项A 、不超过20℃B 、避光并不超过20℃ C 、指2℃~10℃ D 、10℃~30℃ E 、10℃~25℃ 标准答案:B 3 问题: 恒重系指供试品连续两次干燥或炽灼后的重量差小于 答案选项A 、0.5mg B 、0.6mg C 、0.4mg D 、0.3mg E 、0.2mg 标准答案:D 4 问题: “极易溶解”是指溶质1g(ml)能在溶剂()中溶解 答案选项A 、不到1ml B 、1ml C 、10ml D 、100ml E 、1000 ml 标准答案:A 5 问题: 对于原料药,用“含量测定”的药品,其含量限度均用有效物质的百分数(%)表示,此百分数均系指( )百分数 答案选项A 、体积B 、浓度C 、重量D 、容量E 、摩尔 标准答案:C 6 问题: 药物的英文名应尽量采用世界卫生组织制定的 答案选项A 、ChP B 、INN C 、CADN D 、BNF E 、BHP 标准答案:B 7 问题: 《中国药典》中的“精密称定”指称取重量应准确至所取重量的 答案选项A 、±10%B 、百分之一C 、千分之一D 、万分之一E 、百万分之一 标准答案:C 8 问题: 反相高效液相色谱法常用的流动相为 答案选项A 、氯仿B 、丙酮C 、正已烷D 、甲醇-水E 、乙醇-水 标准答案:D 9 问题: 直接酸碱滴定法测定阿司匹林原料含量时,若滴定过程中阿司匹林发生水解反应, 则会使测定结果 答案选项A 、偏高B 、偏低C 、偏高或偏低D 、准确E 、没有影响 标准答案:A 10 问题: 布洛芬中检查的特殊杂质是 答案选项A 、水杨醛B 、水杨酸C 、苯甲酸D 、苯甲醛E 、有关物质 标准答案:E 11 问题: 气相色谱法和高效液相色谱法中常用于鉴别的参数是 答案选项A 、峰高B 、峰宽C 、保留值D 、分离度E 、理论塔板数 标准答案:C 12 问题: 进行药品鉴别试验是为了 答案选项A 、判断已知药物的真伪B 、判断未知药物的真伪 C 、判断药物的纯度 D 、判断药物的优劣 E 、判断药物的有效性 标准答案:A 13 问题: 药品质量标准中性状项下不包括
0832 水分测定法 第一法(费休氏法) 1. 容量滴定法 本法是根据碘和二氧化硫在吡啶和甲醇溶液中与水定量反应的 原理来测定水分。所用仪器应干燥,并能避免空气中水分的侵入;测定应在干燥处进行。 费休氏试液的制备与标定 (1)制备称取碘(置硫酸干燥器内48小时以上)110g,置干燥的具塞锥形瓶(或烧瓶)中,加无水吡啶160ml,注意冷却,振摇至碘全部溶解,加无水甲醇300ml,称定重量,将锥形瓶(或烧瓶)置冰浴中冷却,在避免空气中水分侵入的条件下,通入干燥的二氧化硫至重量增加72g,再加无水甲醇使成1000ml,密塞,摇匀,在暗处放置24小时。 也可以使用稳定的市售费休氏试液。市售的费休氏试液可以是不含吡啶的其他碱化试剂,或不含甲醇的其他伯醇类等制成;也可以是单一的溶液或由两种溶液临用前混合而成。 本试液应遮光,密封,阴凉干燥处保存。临用前应标定滴定度。 (2)标定精密称取纯化水10~30mg,用水分测定仪直接标定;或精密称取纯化水10~30mg,置干燥的具塞锥形瓶中,除另有规定外,加无水甲醇适量,在避免空气中水分侵入的条件下,用费休氏试液滴定至溶液由浅黄色变为红棕色,或用电化学方法[如永停滴定法
(通则0701)等]指示终点;另做空白试验,按下式计算: F=W A?B 式中F为每1ml费休氏试液相当于水的重量,mg; W为称取纯化水的重量,mg; A为滴定所消耗费休氏试液的容积,ml; B为空白所消耗费休氏试液的容积,ml。 测定法精密称取供试品适量(约消耗费休氏试液1~5ml),除另有规定外,溶剂为无水甲醇,用水分测定仪直接测定。或精密称取供试品适量,置干燥的具塞锥形瓶中,加溶剂适量,在不断振摇(或搅拌)下用费休氏试液滴定至溶液由浅黄色变为红棕色,或用永停滴定法(通则0701)指示终点;另做空白试验,按下式计算: ×100% 供试品中水分含量(%)=(A?B)F W 式中A为供试品所消耗费休氏试液的体积,ml; B为空白所消耗费休氏试液的体积,ml; F为每1ml费休氏试液相当于水的重量,mg; W为供试品的重量,mg。 如供试品吸湿性较强,可称取供试品适量置干燥的容器中,密封(可在干燥的隔离箱中操作),精密称定,用干燥的注射器注入适量无水甲醇或其他适宜溶剂,精密称定总重量,振摇使供试品溶解,测定该溶液水分。洗净并烘干容器,精密称定其重量。同时测定溶剂的水分。按下式计算: 供试品中水分含量(%)=W1?W3c1?W1?W2c2 ×100% W2?W3