方法8270 D
气质联用仪测试半挥发性有机化合物
技术翻译:刘金云 mail:piery2006@https://www.doczj.com/doc/0f5559652.html,
1.0范围及应用
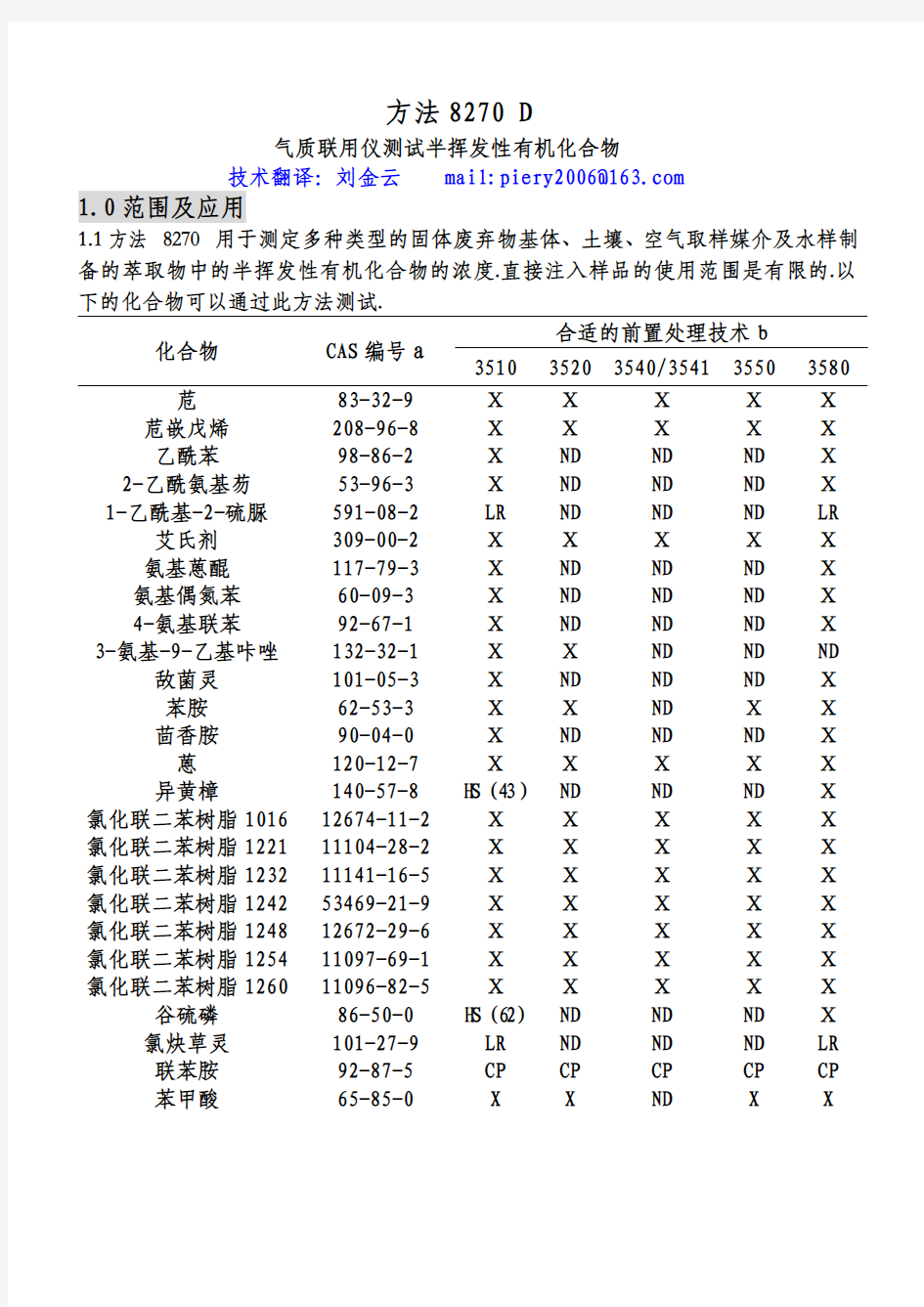
1.1方法 8270 用于测定多种类型的固体废弃物基体、土壤、空气取样媒介及水样制备的萃取物中的半挥发性有机化合物的浓度.直接注入样品的使用范围是有限的.以下的化合物可以通过此方法测试.
合适的前置处理技术b 化合物 CAS编号a
3510 35203540/3541 35503580 苊 83-32-9 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 苊嵌戊烯 208-96-8 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ
乙酰苯 98-86-2 Ⅹ ND ND ND Ⅹ 2-乙酰氨基芴 53-96-3 Ⅹ ND ND ND Ⅹ 1-乙酰基-2-硫脲 591-08-2 LR ND ND ND LR 艾氏剂 309-00-2 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 氨基蒽醌 117-79-3 Ⅹ ND ND ND Ⅹ 氨基偶氮苯 60-09-3 Ⅹ ND ND ND Ⅹ 4-氨基联苯 92-67-1 Ⅹ ND ND ND Ⅹ 3-氨基-9-乙基咔唑 132-32-1 Ⅹ Ⅹ ND ND ND 敌菌灵 101-05-3 Ⅹ ND ND ND Ⅹ
苯胺 62-53-3 Ⅹ Ⅹ ND Ⅹ Ⅹ
茴香胺 90-04-0 Ⅹ ND ND ND Ⅹ 蒽 120-12-7 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 异黄樟 140-57-8 H S(43)ND ND ND Ⅹ 氯化联二苯树脂101612674-11-2 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 氯化联二苯树脂122111104-28-2 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 氯化联二苯树脂123211141-16-5 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 氯化联二苯树脂124253469-21-9 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 氯化联二苯树脂124812672-29-6 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 氯化联二苯树脂125411097-69-1 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 氯化联二苯树脂126011096-82-5 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 谷硫磷 86-50-0 H S(62)ND ND ND Ⅹ 氯炔草灵 101-27-9 LR ND ND ND LR
联苯胺 92-87-5 CP CP CP CP CP
苯甲酸 65-85-0 X X ND X X
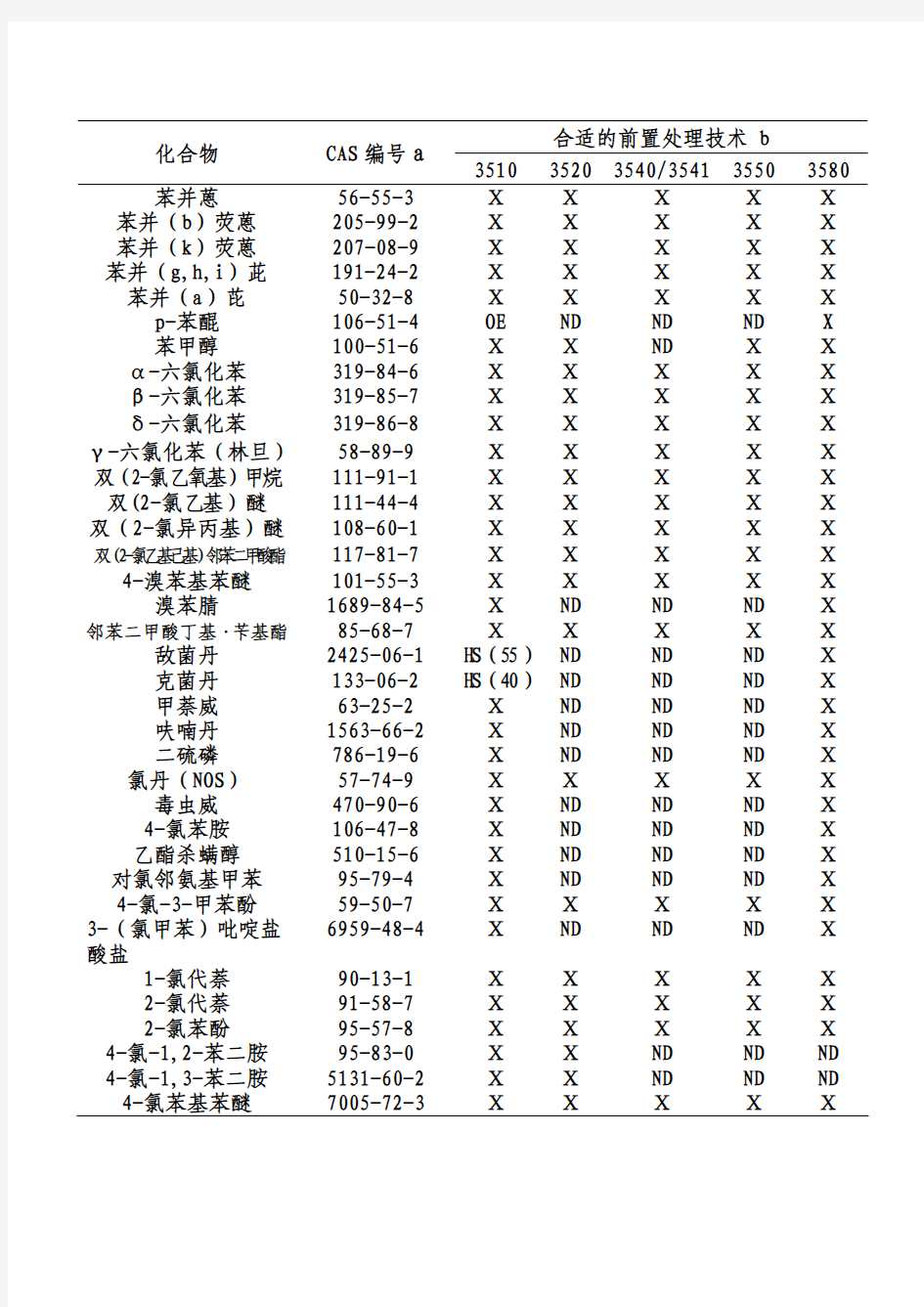
合适的前置处理技术 b 化合物 CAS编号a
3510 35203540/3541 35503580 苯并蒽 56-55-3 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 苯并(b)荧蒽 205-99-2 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 苯并(k)荧蒽 207-08-9 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 苯并(g,h,i)茈 191-24-2 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 苯并(a)芘 50-32-8 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ p-苯醌 106-51-4 OE ND ND ND X
苯甲醇 100-51-6 Ⅹ Ⅹ ND Ⅹ Ⅹ α-六氯化苯 319-84-6 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ β-六氯化苯 319-85-7 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ δ-六氯化苯 319-86-8 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ γ-六氯化苯(林旦)58-89-9 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 双(2-氯乙氧基)甲烷111-91-1 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 双(2-氯乙基)醚 111-44-4 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 双(2-氯异丙基)醚108-60-1 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 双(2-氯乙基己基)邻苯二甲酸酯117-81-7 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 4-溴苯基苯醚 101-55-3 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 溴苯腈 1689-84-5 Ⅹ ND ND ND Ⅹ 邻苯二甲酸丁基·苄基酯85-68-7 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 敌菌丹 2425-06-1 H S(55)ND ND ND Ⅹ
克菌丹 133-06-2 H S(40)ND ND ND Ⅹ
甲萘威 63-25-2 Ⅹ ND ND ND Ⅹ
呋喃丹 1563-66-2 Ⅹ ND ND ND Ⅹ
二硫磷 786-19-6 Ⅹ ND ND ND Ⅹ 氯丹(NOS) 57-74-9 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 毒虫威 470-90-6 Ⅹ ND ND ND Ⅹ 4-氯苯胺 106-47-8 Ⅹ ND ND ND Ⅹ 乙酯杀螨醇 510-15-6 Ⅹ ND ND ND Ⅹ 对氯邻氨基甲苯 95-79-4 Ⅹ ND ND ND Ⅹ 4-氯-3-甲苯酚 59-50-7 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 3-(氯甲苯)吡啶盐6959-48-4 Ⅹ ND ND ND Ⅹ 酸盐
1-氯代萘 90-13-1 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 2-氯代萘 91-58-7 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 2-氯苯酚 95-57-8 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 4-氯-1,2-苯二胺 95-83-0 Ⅹ Ⅹ ND ND ND 4-氯-1,3-苯二胺 5131-60-2 Ⅹ Ⅹ ND ND ND 4-氯苯基苯醚 7005-72-3 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ
合适的前置处理技术 b 化合物 CAS编号a
3510 35203540/3541 35503580 1,2-苯并菲 218-01-9 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 香豆磷 56-72-4 Ⅹ ND ND ND Ⅹ 甲酚定 120-71-8 Ⅹ ND ND ND Ⅹ 丁烯磷 7700-17-6 Ⅹ ND ND ND Ⅹ 2-环己烯-4,6-二硝基-苯酚131-89-5 Ⅹ ND ND ND LR 4,4-DDD 72-54-8 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 4,4-DDE 72-55-9 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 4,4-DDT 50-29-3 Ⅹ Ⅹ Ⅹ Ⅹ X O-异内吸磷 298-03-3 H S(68)ND ND ND Ⅹ S-异内吸磷 126-75-0 Ⅹ ND ND ND Ⅹ 燕麦敌(顺式或反式)2303-16-4 Ⅹ ND ND ND Ⅹ 2,4-二氨基甲苯 95-80-7 D C,0E(42)ND ND ND Ⅹ 二苯并(a,j)吖啶 224-42-0 ⅩND ND NDⅩ二苯并(a,h)蒽 53-70-3 ⅩⅩⅩⅩⅩ二苯并呋喃 132-64-9 ⅩⅩNDⅩⅩ二苯并(a,e)芘 192-65-4 ND ND ND NDⅩ1,2-溴-3-氯丙烷 96-12-8 ⅩⅩND ND ND 苯二甲酸-n-二丁酯 84-74-2 ⅩⅩⅩⅩⅩ二氯萘醌 117-80-6 OE ND ND NDⅩ1,2-二氯代苯 95-50-1 ⅩⅩⅩⅩⅩ1,3-二氯代苯 541-73-1 ⅩⅩⅩⅩⅩ1,4-二氯代苯 106-46-7 ⅩⅩⅩⅩⅩ3,3-二氯联苯胺 91-94-1 ⅩⅩⅩⅩⅩ2,4-二氯苯酚 120-83-2 ⅩⅩⅩⅩⅩ2,6-二氯苯酚 87-65-0 ⅩND ND NDⅩ敌敌畏 62-73-7 ⅩND ND NDⅩ
百治磷 141-66-2 ⅩND ND NDⅩ
狄氏剂 60-57-1 ⅩⅩⅩⅩⅩ酞酸二乙酯 84-66-2 ⅩⅩⅩⅩⅩ乙烯雌酚 56-53-1 AW,ND ND NDⅩ
0S(67)
硫酸二乙酯 64-67-5 LR ND ND ND LR 乐果 60-51-5 H E,ND ND NDⅩ
H S(31)
3,3`-二甲氨基联苯氨 119-90-4 ⅩND ND ND LR 二甲氨基偶氮苯 60-11-7 ⅩND ND ND X 7,12甲基苯并(a)蒽 57-97-6 C P(45)ND ND ND CP
合适的前置处理技术b 化合物 CAS编号a
351035203540/3541 35503580 3,3`-二甲基联苯胺 119-93-7 Ⅹ ND ND ND Ⅹ α,α-二甲基苯乙胺 122-09-8 ND ND ND ND Ⅹ 2,4-二甲苯酚 105-67-9 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 苯二甲酸二甲酯 131-11-3 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 1,2-二硝基苯 528-29-0 Ⅹ ND ND ND X 1,3-二硝基苯 99-65-0 Ⅹ ND ND ND Ⅹ 1,4二硝基苯 100-25-4 H E(14)ND ND ND Ⅹ 4,6-二硝基-2-甲酚 534-52-1 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 2,4-二硝基苯酚 51-28-5 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 2,4-二硝基甲苯 121-14-2 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 2,6二硝基甲苯 606-20-2 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 敌螨普 39300-45-3 CP,HS ND ND ND CP
(28)
二硝丁酚 88-85-7 Ⅹ ND ND ND Ⅹ 二苯胺 122-39-4 Ⅹ Ⅹ X Ⅹ Ⅹ 苯妥英 57-41-0 X ND ND ND Ⅹ 1,2-二苯肼 122-66-7 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 磷酸-n-二辛酯 117-84-0 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 乙拌磷 298-04-4 X ND ND ND Ⅹ 硫丹Ⅰ 959-98-8 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 硫丹Ⅱ 33213-65-9 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 硫酸硫丹 1031-07-8 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 异狄氏剂 72-20-8 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 异狄氏剂醛 7421-93-4 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 异狄氏剂酮 53494-70-5 Ⅹ X ND X Ⅹ 苯硫磷 2104-64-5 Ⅹ ND ND ND Ⅹ 乙硫磷 563-12-2 Ⅹ ND ND ND Ⅹ 氨基甲酸乙酯 51-79-6 D C(28)ND ND ND Ⅹ 乙基甲磺酸 62-50-0 Ⅹ ND ND ND Ⅹ 氨磺磷 52-85-7 Ⅹ ND ND ND Ⅹ 丰索磷 115-90-2 Ⅹ ND ND ND Ⅹ 倍硫磷 55-38-9 Ⅹ ND ND ND Ⅹ 氯氟灵 33245-39-5 Ⅹ ND ND ND Ⅹ
荧蒽 206-44-0 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ
芴 86-73-7 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 2-氟(代)联苯 321-60-8 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 2-氟(代)苯酚 367-12-4 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ
合适的前置处理技术b 化合物 CAS编号a
351035203540/3541 35503580 七氯化茚 76-44-8 X X X X X 七氯环氧化物 1024-57-3 X X X X X 六氯化苯 118-74-1 X X X X X 六氯丁二烯 87-68-3 X X X X X 六氯环戊二烯 77-47-4 X X X X X 六氯乙烷 67-72-1 X X X X X
六氯酚 70-30-4 A W,ND ND ND CP
C P(62)
六氯丙烯 1888-71-7 X ND ND ND X 六甲基磷酸三酰胺 680-31-9 X ND ND ND X 对苯二酚 123-31-9 ND ND ND ND X 茚并(1,2,3-cd)芘 193-39-5 X X X X X 异艾氏剂 465-73-6 X ND ND ND X 异佛尔酮 78-59-1 X X X X X 异黄樟素 120-58-1 D C(46)ND ND ND X
十氯酮 143-50-0 X ND ND ND X
溴苯磷 21609-90-5 X ND ND ND X 马拉硫磷 121-75-5 HS(5)ND ND ND X 顺丁烯二酸酐 108-31-6 HE ND ND ND X 炔雌醇甲醚 72-33-3 X ND ND ND X 噻吩甲吡胺 91-80-5 X ND ND ND X 甲氧氯 72-43-5 X ND ND ND X 甲基胆蒽 56-49-5 X ND ND ND X 4,4`-亚甲基双 101-14-4 O E,O S ND ND ND LR (-2氯苯胺) (0)
4,4`-亚甲基双 101-61-1 X X ND ND ND (N,N-二甲基-苯胺)
甲基甲磺酸 66-27-3 X ND ND ND X 2-甲基萘 91-57-6 X X ND X X 甲基对硫磷 298-00-0 X ND ND ND X 2-甲苯酚 95-48-7 X ND ND ND X 3-甲苯酚 108-39-4 X ND ND ND X 4-甲苯酚 106-44-5 X ND ND ND X
速灭磷 7786-34-7 X ND ND ND X
兹克威 315-18-4 H E,H S ND ND ND X
(68)
灭蚁灵 2385-85-5 X ND ND ND X
久效磷 6923-22-4 HE ND ND ND X
合适的前置处理技术b 化合物 CAS编号a
351035203540/3541 35503580 二溴磷 300-76-5 Ⅹ ND ND ND Ⅹ 萘 91-20-3 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ
萘醌 130-15-4 Ⅹ ND ND ND Ⅹ
1-萘胺 134-32-7 O S(44)ND ND ND Ⅹ
2-萘胺 91-59-8 Ⅹ ND ND ND Ⅹ
烟碱 54-11-5 D E(67)ND ND ND X 5-硝基苊 602-87-9 Ⅹ ND ND ND Ⅹ 2-硝基苯胺 88-74-4 Ⅹ Ⅹ ND Ⅹ Ⅹ 3-硝基苯胺 99-09-2 Ⅹ Ⅹ ND Ⅹ Ⅹ 4-硝基苯胺 100-01-6 Ⅹ Ⅹ ND Ⅹ Ⅹ 2-甲氧基-5-硝基苯胺 99-59-2 Ⅹ ND ND ND Ⅹ 硝基苯 98-95-3 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 4-硝基联苯 92-93-3 Ⅹ ND ND ND Ⅹ 除草醚 1836-75-5 Ⅹ ND ND ND Ⅹ 2-硝基苯酚 88-75-5 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 4-硝基苯酚 100-02-7 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 5-硝基-2-甲苯胺 99-55-8 Ⅹ X ND ND Ⅹ 硝基硅啉-1-氧化物 56-57-5 Ⅹ ND ND ND Ⅹ N-亚硝基二丁胺 924-16-3 X ND ND ND Ⅹ N-亚硝基二乙胺 55-18-5 X ND ND ND Ⅹ N-亚硝基二甲胺 62-75-9 Ⅹ X X X Ⅹ N-亚硝基甲基乙胺 10595-95-6 Ⅹ ND ND ND Ⅹ N-亚硝基二苯胺 86-30-6 Ⅹ X X X Ⅹ N-亚硝基二丙胺 621-64-7 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 亚硝基吗啉 59-89-2 ND ND ND ND Ⅹ N-亚硝基呱啶 100-75-4 Ⅹ ND ND ND Ⅹ 亚硝基吡咯烷 930-55-2 Ⅹ ND ND ND Ⅹ 八甲基焦磷酰胺 152-16-9 LR ND ND ND LR 4,4`-羟基二苯胺 101-80-4 Ⅹ ND ND ND Ⅹ 对硫磷 56-38-2 Ⅹ X ND ND Ⅹ
五氯苯 608-93-5 X ND ND ND X 五氯硝基苯 82-68-8 Ⅹ ND ND ND Ⅹ 五氯苯酚 87-86-5 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 非那西丁 62-44-2 Ⅹ ND ND NDⅩ 菲 85-01-8 Ⅹ Ⅹ X X X 菲巴比妥 50-06-6 Ⅹ ND ND ND Ⅹ 苯酚 108-95-2 D C(28)Ⅹ Ⅹ Ⅹ Ⅹ
合适的前置处理技术b 化合物 CAS编号a
3510 35203540/3541 35503580 1,4-苯二胺 106-50-3 Ⅹ ND ND ND Ⅹ 甲拌磷 298-02-2 Ⅹ Ⅹ Ⅹ Ⅹ Ⅹ 伏杀硫磷 2310-17-0 HS(65)ND ND ND Ⅹ 亚胺硫磷 732-11-6 HS(15)ND ND ND Ⅹ 磷胺 13171-21-6 H E(63)ND ND ND Ⅹ
酞酐 85-44-9 C P,H E(1)ND ND ND CP 2-皮考啉(2-甲基吡啶)109-06-8 Ⅹ X ND ND ND 胡椒基亚砜 120-62-7 Ⅹ ND ND ND Ⅹ 拿草特 23950-58-5 Ⅹ ND ND ND Ⅹ 丙硫氧密啶 51-52-5 LR ND ND ND LR 芘 129-00-0 Ⅹ X X X Ⅹ 间苯二酚 108-46-3 DC,OE ND ND ND Ⅹ
(10)Ⅹ 黄樟素 94-59-7 Ⅹ ND ND ND Ⅹ 番木鳖碱 57-24-9 AW,0S ND ND ND Ⅹ
(55)
菜草特 95-06-7 Ⅹ ND ND ND Ⅹ
叔丁磷 13071-79-9 Ⅹ ND ND ND Ⅹ 1,2,4,5-四氯苯 95-94-3 X ND ND ND Ⅹ 2,3,4,6-四氯苯酚 58-90-2 X ND ND ND Ⅹ 杀虫畏 961-11-5 Ⅹ ND ND ND Ⅹ 四乙基二硫代焦磷酸3689-24-5 Ⅹ X ND ND ND 焦磷酸四乙酸酯 107-49-3 Ⅹ ND ND ND Ⅹ 硫磷嗪 297-97-2 Ⅹ ND ND ND Ⅹ 苯硫酚(硫酚) 108-98-5 X ND ND ND Ⅹ 甲苯二异氰酸酯 584-84-9 HE(6)ND ND ND Ⅹ 邻甲苯胺 95-53-4 Ⅹ ND ND ND Ⅹ
毒杀芬 8001-35-2 X X X X X 1,2,3-三氯苯 120-82-1 Ⅹ X X X Ⅹ 2,4,5-三氯苯酚 95-95-4 Ⅹ X ND X Ⅹ 2,4,6-三氯苯酚 88-06-2 X X X X X 氟乐灵 1582-09-8 Ⅹ ND ND ND Ⅹ 2,4,5-三甲基苯胺 137-17-7 Ⅹ ND ND ND Ⅹ 磷酸三甲酯 512-56-1 H E(60)ND ND NDⅩ 1,3,5-三硝基苯 99-35-4 Ⅹ ND ND ND X 磷酸三(2,3-二溴丙基酯)126-72-7 Ⅹ ND ND ND LR
合适的前置处理技术b 化合物 CAS编号a
351035203540/3541 35503580 磷酸邻三甲苯酯 78-32-0 Ⅹ ND ND ND Ⅹ O,O,O-三乙基代磷酸酯126-68-1 Ⅹ ND ND ND Ⅹ a: CAS-化学文摘服务处.
b: 其它可以接受的前置处理方法参见第1.2节.
分析物目录要点:
AW=在萃取和储存时,吸附到玻璃器皿内壁上.
CP=不可重复的色谱性能
DC=不理想的分布系数(括号中的数字为回收率百分数)
HE=萃取时的水解作用在酸性或碱性条件的催化下加快(括号中的数字为回收率百分数).
HS=储存时的水解作用(括号中的数字为稳定性百分率)
LR=较低的回应
ND=没有测试到
OE=萃取时,在碱性条件的催化作用下氧化作用加快(括号中的数字为回收率百分数).
OS=储存时被氧化(括号中的数字为稳定性百分率).
X=通过此种技术回收率大于70%.
1.2除了在以上的分析物目录中列出的样品制备方法外,方法3535描述的是一个固相萃取程序,可应用于从TCLP浸出液中(表16与表17中包含性能数据)提取半挥发性物质.方法3542描述的是气体中的半挥发性有机化合物的样品制备程序,气体是通过方法0010(表11中含有代用物的性能数据)取样的.方法3545描述的是利用溶剂从固体中自动萃取半挥发性物质的装置(表12包含有性能数据).方法3561描述的是从固体中萃取多环芳香烃类(PAHs)的超临界流体装置(见表格13,14,15中的性能数据). 1.3对于能溶解于二氯甲烷的中性、酸性和碱性有机化合物,且它们能够被洗脱,无需衍生化便可从气相色谱熔融二氧化硅毛细管柱(柱上涂有少量的极性硅酮)显现尖锐的峰,大多数这样的有机化合物可用方法8270来定量测试.这些化合物包括:多环芳香烃类(PAHs)、氯化烃类、农药、邻苯二甲酸酯类、有机磷酸酯类、亚硝胺类、卤代醚类、醛类、醚类、酮类、苯胺类、吡啶类、喹啉类、芳香族硝基化合物、酚类(包括硝基酚类)。表1中列出了这些化合物的名称及其已经被评估过的特径离子. 在大多数情况下,方法8270不适合于多元分析物的量化.例如:氯化联苯树脂,毒杀芬,氯丹等,因为对于这些分析物,此方法的灵敏度有限.当萃取物中的浓度达到要求时,方法8270可用来证明这些分析物的存在.对于多元分析物如氯化联苯树脂、毒杀芬、氯丹的校正和量化,请参考方法8081及方法8082作为指导.
1.4下列化合物在使用方法8270测定时,需经过特别处理:
1.4.1联苯胺在溶剂浓缩时可能会发生氧化反应而损失,其色谱特性也较差.
1.4.2在碱性的条件下,从含水基质中萃取α-六氯化苯,γ-六氯化苯硫丹Ⅰ,硫丹Ⅱ,异狄氏剂时,会发生分解.如果估计含有这些化合物,应当在中性的条件下萃取.
1.4.3六氯环戊二烯在气相色谱仪的入口处会发生热分解,在丙酮溶液中会发生化学反应及光化学分解.
1.4.4 N-亚硝基二甲胺在所述的色谱条件下很难从溶液中分离.
1.4.5 N-亚硝基二苯胺在气相色谱仪的入口分解,并且不能从二苯胺中分离出来.
1.4.6五氯苯酚,2,4-二硝基苯酚,4-硝基苯酚,苯甲酸,4,6-二硝基-2-甲酚,4-氯-3-甲酚,硝基苯胺,4-氯苯胺,苯甲醇的色谱特性不稳定,特别是在GC的系统被高沸点的材料污染之后.
1.4.7吡啶在本方法中列出的GC入射口温度条件下,其色谱特性差,降低入射口的温度可能会减少降解的量.然而,分析者在调节注射口的温度时应引起注意,其它的分析物的性能可能相反地受到影响.因此,当吡啶同其它的目标分析物一起需要分析时,可能必要进行不同的分析过程.此外,吡啶在样品萃取物进行蒸馏浓缩时可能会损失掉.因此,在操作浓缩步骤时要特别细心,否则以上列出的许多萃取方法可能会产生较低的回收率.鉴于这个原因,分析者可以考虑使用其它的萃取技术如加压流体萃取(方法3545)或超临界流体萃取.超临界流体萃取需要的萃取物体积小,因此,在很多的情况下,它减少了或消除了蒸馏浓缩技术的必要.
1.4.8甲苯二异氰酸酯在水中会迅速地水解(半存留期小于30分钟).因此,从含水基质回收这种化合物是不可期望的.另外,在固体基质中,甲苯二异氰酸酯经常同乙醇和胺反应产生会产生氨基甲酸酯.因此,它同含有这些物质的溶剂不能共存.
1.4.9当存在由样品的制备和/或色谱问题产生的局限时,以上分析列里的分析物会变衰弱.
1.5对于土壤及沉积物样品,方法8270测试单种化合物的评估量化限度(EQL)大约是660μg/kg (湿重);废弃物的EQL为1-200㎎/kg(取决于基质及制备方法);地下水样品为10μg/kg,(见表2).对于为了防止检测器饱和而需要稀释的样品,其EQL也会相对较高.
1.6本方法仅限于有GC-MS使用经验并精通质谱解读的分析者,或在此类分析者的指导下进行.每个分析者都必须展示他能够利用此方法得到可接受的结果。
2.0方法概述
2.1使用气质联用技术分析的样品在测试前需使用合适的样品制备方法(参见方法3500),当有必要时,使用样品提纯程序提纯(参见方法3600).
2.2通过将样品萃取物注入至有很窄的小孔的熔融的二氧化硅毛细管柱中,使半挥发性化合物导入至GC/MS中.气相色谱柱使用温度程控, 使分析物分离,然后通过连接到气相色谱仪上的质谱仪分析.
2.3经毛细管柱洗脱的分析物通过喷射式或直接连接的方式导入质谱式中.目标分析物的辨别是利用真实标准的电子冲击(或类似于电子冲击)谱比较它们的质谱来完成的.量化是通过比较主要(量化)离子的回应来完成,此主要离子与使用五点校正曲线的内标法相关.
2.4本方法包含特殊的校正及质量控制步骤,取代了方法8000提供的一般性要求.
3.0干扰
来自所有空白,样品及掺料溶液的原始GC/MS数据必须评估是否存在干扰.确定干扰是否来源于样品制备和/或提纯程序并采取矫正措施消除问题产生的原因.
3.2当高浓度的样品和低浓度的样品连续地分析时,会产生转移污染.为了减少样品的转移,在样品的注射之间隙必须使用溶剂冲洗样品的注射器.当遇到一个非常高浓度的样品时,在分析完之后,需接着分析溶剂,以确认是否存在交叉污染.
4.0仪器及材料
4.1气相色谱-质谱联用系统
4.1.1气相色谱仪-一个以温度编程的分析系统,此气相色谱仪适合不分流进样.所有需要的附属设备,包括注射器,分析柱及气体.毛细管柱与气源是直接连接在一起的.
4.1.2柱-30-m×0.25mm的内径(或内径为0.32mm)的熔融二氧化硅毛细管柱(J&W Scientific DB-5型或同级品),涂有1μm厚的薄膜硅硐.
4.1.3质谱仪
4.1.3.1在1秒钟内能从原子质量为35扫描到500. 使用以电子轰击离子化模式获得的70V(参考值)电子能量.当1μL的GC/MS的调试标准溶液(50ng的十氟三苯基磷)注入GC后,质谱仪必须能产生符合表3标准的质谱图.
4.1.3.2如果离子阱质谱计能够进行轴向调制以减少离子-分子反应,并且能够产生符合EPA和NIST图书馆的类电子冲击谱,则也可以使用此质谱计。当5ng或50ng 的DFTPP加入时,质谱仪必须能够产生符合标准的质谱图,此标准在表3中有列出。
4.1.4GC/MS接口-对GC与MS可能使用的任何接口,在每次注入50ng时,能为每种目标化合物提供可接受的校正点,且能够达到可接受的调谐性能标准.对于小孔的毛细管柱,接口是经常同毛细管直接导入质谱源.
4.1.5数据系统-质谱仪与计算机系统连接在一起。计算机系统应能够连续地获取并储存在整个的色谱项目中获得质谱图的机器可读媒介。计算机的软件能够为某种原子量的离子搜索GC/MS数据文件,并能够将这种离子丰度以时间或扫描数目为横坐标描点。这种类型的点叫做“当前提取离子曲线”(EICP)。软件还可以将某段时间或扫描数目限度内的EICP丰度的点连接起来。最新版本的EPA及NISI质谱库也应该取得。
4.1.6保护柱(供选择)-(J&W减活的熔融二氧化硅,0.25-mm内径×6m,或同级品)在注射口和连接柱接头的分析柱之间(Hewlett-Packard 产品目录号5062-3556或同级品).
4.2注射器-10μL.
4.3容量瓶,A级,合适的大小,带有磨口玻璃瓶塞.
4.4分析天平-能够称量0.0001g.
4.5试剂瓶-玻璃瓶,有聚四氟乙烯(PTFE)边缘的螺纹盖或有卷曲顶端.
5.0试剂
所有测试用到的无机化学药品都必须是试剂等级. 除非有特别说明,所有的试剂都必须符合美国化学协会分析试剂委员会的说明,在那可获得此规格说明.其它等级的化学品也可以使用,只要首先已探明其纯度足够高,能达到使用时不降低测试的准确度的效果.
5.2不含有机物的试剂水.本方法中提到的水都是不含有机物的试剂水,见本手册第一章的定义.
5.3备用标准溶液(1000μg/L)-标准溶液可以通过纯的标准物质制备或购买已经标定的溶液.
5.3.1准确称量约0.100g纯物质制备备用标准溶液.将此物质溶解于农药等级的丙酮中或其它合适的溶剂中,并将它在10mL的容量瓶中稀释至刻度线.为了分析者的方便,可以使用更多体积的溶液.当化合物的纯度经鉴定达到96%的纯度或以上时,此物质的重量在计算备用标准溶液的浓度时,勿需作修正.市场上配制的任保浓度的备用都可以使用,只要是经过生产商或独立机构的检测.
5.3.2将备用标准溶液转移至带有PTFE边缘螺纹塞的瓶中,并置于-10℃或低于-10℃的黑暗环境下保存,或根据标准生产商建议的方法储存备用溶液.备用标准溶液必须定期检验,防止分解或挥发,特别是在利用它们配备校正标准溶液之前.
5.3.3备用标准溶液必须在1年内或更短的时期内被替换掉,当它同质量控制样品作比较显示此溶液有问题时.
5.3.4建议亚硝基胺应单独放置在一种校正混合溶液中,而不要将它同其它的校正混合溶液混合.当使用预先混合的鉴定标准溶液时,请参考生产商的指导.
5.3.5当溶液同盐酸的盐混合时可能会产生盐酸,这会增加分析的难度.当使用预先鉴定的标准溶液时,请参考生产商的指导.
5.4内标溶液-推荐使用1,4-二氯代苯-d4、萘-d8、苊-d10、菲-d10、1,2-苯并菲-d12和二萘嵌苯-d12 作为内标物质.其它满足第7.3.2中说明的化合物也可以作为内标物质.
5.4.1取每种化合物0.200g用少量的二硫化碳溶解.将溶液转移至50mL的容量瓶中并用二氯甲烷稀释至体积,最终溶剂中含大约20%的二硫化碳.大多数的化合物能溶解
于少量体积的甲醇,丙酮或甲苯中,但二萘嵌苯-d12除外.最终的溶液含每种标准物质的浓度为4,000ng/μL。每1mL进行分析样品萃取物应掺入10μL的内标溶液,产生的内标物浓度相当于40ng/μL。当不使用时,应置于-10℃或-10℃以下的环境中保存。当使用预先混合的鉴定溶液时,合适的放置时间和储存温度应根据生产商文件的要求。
5.4.2为了达到更低的检测限而使用更灵敏的质谱仪时,可能需要更稀释的内标溶液。内标物质峰的面积应为处于中点校正分析的目标分析物面积的50-200%之间。
5.5GC/MS调试标准溶液-配制一个含有十氟三苯基磷(DFTPP)浓度为50ng/μL的二氯甲烷溶液。此标准溶液中也含有4,4`-DDT,五氯苯酚,联苯胺,浓度也都为50ng/μL,以检验注射口的惰性及时性气相色谱柱的性能。当不使用时,应放于-10℃或-10℃以下的环境中保存。当为了达到更低的检测限而使用更灵敏的质谱仪时,可能需要更稀释的调试溶液。当使用预先混合的鉴定溶液时,合适的放置时间和储存温度应根据生产商文件中的要求。
5.6校准标准溶液-至少配制浓度不同的5个校准标准溶液.至少有一种校准标准溶液有浓度等于或低于样品的浓度,以符合本项目数据质量目标的要求.另外的标准溶液浓度范围应与实际样品的浓度范围一致,但不要超过GC/MS系统的工作范围.每一标准溶液中都含有使用本方法检测的每种分析物.
5.6.1EPA目的是要求每种分析的校正标准溶液中含有所有的目标分析物,这些目标分析物并不包含整个分析物目录(第1.1节)中的物质,分析物目录中的物质已经本方法证明有效.然而,实验室却不能报告校准标准溶液中不含有物质的定量分析结果. 5.6.2在分析之前,每1mL的校准标准溶液中应掺入10μL的内标溶液.所有的标准溶液都必须置于-10℃或以下保存,并一年配制一次,或当检验标准溶液发现问题时,应立配制.校正验证标准溶液应每周配制并于4℃下储存. 当使用预先混合的鉴定溶液时,合适的放置时间和储存温度应根据生产商文件中的要求。
5.7代用标准溶液-推荐的代用物质为为苯酚-d6,2-氟代苯酚,2,4,6-三溴苯酚,硝基苯-d5,2-氟代苯及对三联苯-d14,配制代用溶液请参考方法3500作为指导。
5.7.1代用标准溶液的检验-在萃取,提纯,浓缩步骤后确定空白萃取物的合适浓度,将此浓度溶液注入至GC/MS测试代用标准物质的回收率。当配制新的代用掺料溶液时,建议采取此检验步骤。
备注:方法3561(PAHs的超临界流体萃取)建议使用溴苯及对四联苯,以便更好地将此方法中列出的PAHs覆盖在范围内。
5.7.2为了达到更低的检测限而使用更灵敏的质谱仪时,可能需要更稀释的代用溶液。
5.8基体掺料及实验室控制标准溶液-配制基体掺料标准溶液请参考方法3500作为指
导,相同的标准溶液可用来作实验室控制标准溶液(LCS)。
5.8.1基体掺料的检验-在所有的萃取,提纯,浓缩步骤之后确定空白萃取物中最合适的基体掺料浓度,将此浓度溶液注入至GC/MS测试回收率。当配制新的基体掺料溶液时,建议采取此检验步骤。
5.8.2为了达到更低的检测限而使用更灵敏的质谱仪时,可能需要更稀释的基质掺料溶液及实验室控制标准掺料溶液。
5.8.3某些实验项目可能需要特殊的化合物掺料,因为方法3500中列出的掺料化合物并不代表本项目需要的目标化合物。当这种情况发生时,基体掺料标准溶液和实验室控制标准溶液应在甲醇中配制,溶液中每种化合物的浓度适合此分析项目。
5.9溶剂-丙酮,己烷,二氯甲烷,异辛烷,二硫化碳,甲苯或其它合适的溶剂。所有的溶剂都必须是符合农药等级或同级品。
6.0样品的收集,储存和处理
6.1见《有机分析物》一章第4.1节的介绍性内容。
6.2将样品的萃取物于-10℃置于黑暗的环境下保存。萃取物装在密封的指瓶中(例如,螺纹盖指瓶或卷顶指瓶,指瓶上的PTFE-线型隔膜未被刺穿。
7.0程序
7.1在执行GC/MS分析之前,样品一般使用以下前置处理方法中的一种进行处理。基体 前置处理方法
气体(颗粒状及吸附树脂) 3542
水(包括TCLP沥取液) 3510,3520,3535
土壤和沉积物 3540,3541,3545,3550,3560,3561 废弃物 3540,3541,3545,3550,3560,3561,
3580
7.1.2在非常有限的应用当中,才可以将样品直接地用10-μL的注射器直接注入到GC/MS系统中。而此直接注入法的检测限很高(大约为10,000μg/L)。因此,只有在估计浓度值超过10,000μg/L时才允许这种做法。
7.2萃取物质提纯-在利用GC/MS分析之前,萃取物可使用如下方法中的一种进行提纯。
目标分析物 方法
苯胺类及苯胺的衍生物 3620
酚类 3630,3640,8041a
邻苯二甲酸酯类 3610,3620,3640
亚硝基胺类 3610,3620,3640
有机氯农药及多氯联苯(PCBs) 3610,3620,3630,3660,3665
硝基芳烃类及环酮类 3620,3640
多环芳香烃类(PAHs) 3611,3630,3640
卤代醚类 3620,3640
氯化烃类 3620,3640
有机磷农药类 3620
石油的废弃物 3611,3650
所有的碱性,中性及酸性优先污染物 3640
如果在气相色谱/火焰离子化检测器遇到干扰时,方法8041中包含衍生化技术和气相色谱/电子捕获检测器分析技术。
7.3初始校正
使用以下的建议作为指导,设置GC/MS的运行条件。
品质范围 35-500amu(原子质量单位)
扫描时间 1秒/每次扫描
初始温度 40℃,保持4分钟
温度项目 40-270℃,每分升温10℃。
最终温度 270℃,保持此温度直到苯并(g,h,i)芘被洗脱出来注射器温度 250-300℃
输送线温度 250-300℃
离子源温度 根据制造商的说明书
注射器 Grob型,不分流式
注射体积 1-2μL
载气 氢气流速为50cm/秒,氦气为30cm/秒
只适用于离子阱质谱仪: 根据制造商的建议设置轴向调谐器,管道温度和放射
电流.
如果质谱仪的灵敏度足够高,允许使用分流式注射器.
7.3.1GC/MS系统在注入50ng的DFTPP时,必须能够使用硬件调谐.只有在调谐标准满足之后才可以开始分析.
7.3.1.1在仪器的供货商未提供如何获取DFTPP质谱图的相关建议的情况下,以下提供的方法可能有用:将获得的3次扫描(峰的顶点扫描,取代峰顶及跟随峰顶的扫描)平均。需要减去背景,这一步骤在DFTPP洗脱之前由不超过20次扫描的单次扫描来完成。减去背景的目的仅仅是为了消除掉柱渗透或仪器的背景离子。不要将DFTPP峰的一部分减去。
7.3.1.2在调制允收的标准时,请使用表3中的DFTPP原子质量强度标准。另外,其它调谐标准檔(例如,CLP,方法525或是制造商的说明书)也可以合使用,只要是方法性能不会受到影响。
备注:所有后续与DFTPP分析相关的标准溶液,样品,MS/MSDs及空白溶液都必须
使用相同的质谱仪器条件。
7.3.1.3GC/MS的调试溶液也用来评估气相色谱柱的性能和注射口的惰性,DDT降解成DDE及DDD的量不得超过20%(分解百分率的计算参考方法8081第8.0节).iv 当联苯胺和五氯苯酚有正常响应时,它们应该存在。不能看见有峰的拖尾。
7.3.1.4如果降解过多和/或看见的色谱图很差,注射口可能需要清洗,同时还可能有必要将开始一段6-12英寸的毛细管柱折断。在注射口与分析柱之间使用保护柱(见第4.1.6节)可以延长毛细管柱的性能。
7.3.2第5.4节选择的内标溶液应允许色谱图中的大多数目标成份相对于其中一种内标溶液有0.80-1.20的延滞时间。使用相关内标溶液的标准(基)峰离子作为量化时的主要离子(见表1)。如果观察到有干扰,使用下一个最强烈的离子作为量化离子(例如,对于1,4-二氯代苯-d4,使用152 m/z作为量化)。
7.3.3分析每种校准标准溶液(包括内标溶液)1-2μL,并将每一目标分析物(如表1)浓度对应的主要特征离子的面积制成表格。至少需要五个校准标准溶液(见第5.6节及方法8000)。对于所有的标准溶液及样品萃取物的注入体积都是一样。图1中显示的是含有碱性,中性及酸性分析物的校准标准溶液的色谱图。
内标溶液其相关的目标分析物响应系数(RF)计算公式如下:
RF=A S×C IS/A IS×C S
上式中:
A S=分析物或代用物质的峰面积(或高度)
A IS=内标物的峰面积(或高度).
C S=分析物或代用物质的浓度,单位为μg/L.
C IS=内标物质浓度, 单位为μg/L.
7.3.4系统性能检验化合物(SPCCs)
7.3.4.1在使用标准曲线之前,必须使用系统性能检验的化合物进行检验,以确保符合最小的平均响应系数,对于半挥发性有机物,检验系统性能的化合物(SPCCs)为:N-亚硝基二丙胺;六氯环戊二烯;2,4-二硝基苯酚及4-硝基酚。
7.3.4.2这些化合物的最小可接受的平均响应系数值为0.050。这些检验系统性能化合物一般具有很低的响应系数(0.1-0.2),当色谱系统变差或标准物质变差时,其响应都会减小。它们往往是最先显示性能不好。因此,当系统校正时,这些化合物必须满足此最小值要求。
7.3.4.3如果没有满足最低响应系数要求,必须对系统作评估。在样品分析开始之前必须采取矫正措施。可能存在的问题包括标准混合物的降解,注射入口处被污染,分析柱前端部分的污染及毛细管柱中或色谱系统中某些位置可以活动。在样品的分析开始之前,必须满足检验化合物最低响应系数的要求。
7.3.5校正检验化合物(CCCs)
7.3.5.1校正检验化合物(CCCs)的目的是从系统统一性的立场去评估校准。这引起检验校准化合物的高度变化性可能表明系统产生泄漏或柱中某些地方产生反应。而满足校准检验化合物标准不可代替代方法8000中第7.0节的一种方法去成功地校准目标分析物。
7.3.5.2根据每种目标分析物的响应系数,计算它的平均响应系数及相对标准差(RSD)。每种目标分析物的相对标准差应小于或等到于15%,然而每一个校准检验化合物(CCCs)的相对标准差必须小于或等到于30%。
n
平均RF=∑RF i /n 标准差
相对标准差=SD/RF×100
7.3.5.3如果任保校准检验化合物的相对标准差大于30%,那么,色谱系统发生过多的反应而不适合作分析。清洁或更换注射器衬垫和/或毛细管柱,然后重复第7.3节开始的校正程序。
7.3.5.4如果某分析项目中校准检验化合物不包含在分析物目录中,那么,请不要将此化合物包含在校准标准物质中,然后参见方法8000第7.0节.
7.3.6评估保留时间-每种校准标准物质的每一目标分析物的相对保留时间应符合在0.06个单位的相对保留时间内,其后洗脱的目标分析物一般都能更好地符合此标准.
分析物的保留时间
相对保留时间(RRT)=内标物的保留时间
7.3.7目标分析物的线性-如果某目标分析物的相对标准差小于或等于15%.则可假定它的相对回应系数在校准范围内恒定的.必须使用它的平均响应系数来量化(第7.6.2节).
7.3.7.1如果某目标分析物的相对标准差大于15%,另外选择的校准方法请参照方法8000第7.0节.在此种情况下,必须选择其中一种方法对GC/MS系统进行校正,或是进行一种新的始校正.
备注:方法8000的线性标准目标值是20%的相对标准差.此标准适用于GC和HPLC方法而不是GC/MS.方法8270要求达到15%的标准差,以证明有足够的线性度使用平均响应系数.
7.3.7.2当相对标准差超过15%时,通过描点并目视分析标准曲线能够有效地发现问题.分析标准曲线能够发现分析中产生的问题,包括标准溶液配制中产生的错误,色谱柱中存在的活动的部位,色谱特征较差的分析物,等等.
7.4GC/MS校正验证-校正验证由三个步骤构成,在每12小时的分析班次开始时都需
要执行此步骤.
7.4.1在分析样品或校准标准溶液之前,将50ng的DFTPP标准溶液注入至GC/MS系统中.在样品分析开始之前,产生的DFTPP质谱图必须符合表3中的标准.这些标准都必须每12小时的样品被分析班次得到证明.
7.4.2在样品分析之前,每种目标化合物的初始校正(第7.3节)必须每12小时验证1次.验证时使用样品所使用的介绍性技术及条件.这是通过分析处于GC/MS校正范围中点浓度的校正标准溶液来完成.分析校正标准溶液的结果必须符合第7.4.4-7.4.7所提供的验证允收标准.
备注:DFTPP和校准验证标准溶液可混合至一种标准溶液中,只要符合本项目的调谐及校正验证允收标准而没有干扰.
7.4.3方法空白可以在校准标准溶液之后或是在整个分析班次中的任何时间分析,以确保整个系统(导入系统,传输线路及GC/MS系统无污染.如果分析方法空白显示有污染,可以接着分析一个溶剂空白以证明污染不是来自上个标准溶液或样品.参考方法8000B的方法空白性能标准.
7.4.4系统性能检验化合物(SPCCs)
7.4.4.1每12小时的分析批次必须使用系统性能检验化合物对系统性能进行检验。校准验证标准溶液每一系统性能检验化合物必须符合最小响应系数为0.050的标准。此检验方法同初始校准使用的检验方法相同。
7.4.4.2如果没有满足最小响应系数的要求,必须对系统作评估,在样品的分析开始之前必须采取必要的矫正措施。问题产生的可能原因包括标准混合物质的降解,注射器入口处被污染,分析柱的前端部分被污染,柱中或色谱系统中存在活动位置。在样品开始分析之前,必须满足此检验要求。
7.4.5校正检验化合物(CCCs)
7.4.5.1在系统的性能满足之后,使用表4中列出的校正检验化合物来检验初始校正的有效性.当执行平均响应系数模式校正时采用差异百分率来表示.而当使用回归调准模式时则采用漂移百分率来表示,计算差异百分率及漂移百分率参见方法8000第7.0节.
7.4.5.2如果每种校正检验化合物的差异百分率小于或等于20%,可以设想初始校正是有效的.如果有一种校正检验化合物没有满足此标准(例如,差异百分率或漂移百分率大于20%),则在开始分析样品之前必须采取矫正措施.如果某项目的分析物目录中没有校正检验化合物,因此,也不包含有校正标准溶液当中,则所有的分析物都必须满足20%的差异百分率及漂移百分率的标准.
7.4.5.3与系统性能检验化合物下面列出的相似问题也同样会影响到校正检验化合物.如果问题不能采取其它的措施校正.必须进行新的初始校正.在分析样品之前必须满
足校正检验化合物的标准.
7.4.6内标物保留时间- 在获取数据后或区划获取数据时,必须对校正验证标准物质中的内标物的保留时间立即作出评估.如果从最近的初始校正的中点标准浓度开始的任保内标物的保留时间变化超过30秒,则应检查是否色谱系统有故障并采取必要的矫正措施。当采取矫正措施过后,所有的在系统故障期间分析的样品都必需重新分析。
7.4.7内标回应-如果从最近的初始校正的中点标准浓度开始,校正验证标准物质中的任何内标物的萃取离子电流曲图(EICP)的面积变化超过达到两位数以上(-50%到+100%),则必须检查质谱仪是否有故障并采取合适的矫正措施。采取矫正措施过后,所有的在系统故障期间分析的样品都必需重新分析。
7.5气质联用仪(GC/MS)分析样品
强烈建议用GC/FID或GC/PID进行鉴别的样品萃取物使用与GC/MS系统相同型号的毛细管柱。这将有助于减少非常高浓度的有机化合物对GC/MS系统的污染。
7.5.2将样品萃取物预热至室温。在分析刚开始之前,向1mL从样品品前置处理获得的浓样品萃取物中加入10 μL内标溶液。
7.5.3注入1-2 μL样品萃取物至GC/MS系统中,使用与校正(第7.3节)相同的操作条件。除非使用的GC/MS系统灵敏度更高,否则注入的体积中应含有100ng碱性或中性及200ng的酸性代用物质(假定回收率为100%),且代用溶液的浓度低于第5.7节所列出的浓度。注入的体积必须与校正标准溶液所使用的相同。
7.5.4如果任何量化离子的响应超过GC/MS系统的初始校正范围,样品萃取物必须稀释并重新分析。稀释的萃取物中应另外加入内标溶液以维持和校正标准溶液(40ng/μL,除非使用的GC/MS系统的屡敏度非常高)中相等的浓度。当样品干扰到主要离子时,才会使用次级离子量化。
备注:监控内标物保留时间及所有的样品、掺料、空白及标准溶液的响应因子(面积计数)是有用的诊断工具,可以有效地检查到漂移的方法性能,很差的注射运行,并可预料到系统检查和/或维修的需求。
7.5.4.1当样品中化合物的离子使检测器饱和时,这类分析必须接着分析一个由干净的溶剂配制的仪器空白。如果分析空白中存在干扰,那么系统一定被污染了。直到空白分析不含有干扰,否则样品分析不可继续进行。
7.5.4.2所有的稀释溶液的主要成分(以前饱和的峰)的响应处于曲线线性范围的上半部分。
7.5.5对于所需要的检测限处于电子轰击质谱仪的正常范围以下的应用,使用选取离子监控是可以接受的。然而。选择离子监控在化合物识别时提供的可信度不高,除非是每一种化合物的多种离子得到监控。
7.6定性分析
7.6.1通过本方法定性鉴别化合物是基于保留时间和在校正之后,比较样品的质谱图与特征离子在参考的质谱图库中的质谱图。实验室必须使用本方法相同的条件去产生质谱图库。参考质谱图库中的特征离子被定义为相对强度最大的三个离子,或是当参考质谱图中小于3个离子时,定义为相对强度超过30%的离子。当符合以下标准时,化合物得以鉴别。
7.6.1.1通过数据系统的目标化合物搜索途径选择色谱峰。搜索的依据是在目标化合物处于化合物的独有的保留时间,存在离子固有的一个目标色谱峰。选择这种色谱峰是符合此标准的。
7.6.1.2样品成份的相对保留时间是处于标准成分保留时间的±0。06个单位内。特征离子的相对强度与参考谱库中的特征离子的相对强度相差不超过30%(例如,对参考质谱库中丰度为50%的特征离子,样质量谱图中的范围是从20%-80%。
7.6.1.4结构同分异构体会产生非常相似的质谱图,如果它们GC保留时间的差别很大,则应把它们当作不同的同分异构体。如果此两种同分异构体的两峰之间谷的高度小于两峰总高度的25%,GC可以达到足够的分辨率。另外,同分异构体被看作是同分异构对。非对映的同分异构对(例如,异黄樟与异黄樟素)可以通过GC分离,并通过GC鉴别,量化及报告两种化合物的总量。
7.6.1.5当样品的成分从色谱上不可分辨并产生不止一种分析物的离子产生的质谱图时,鉴别受到阻碍。当气相色谱峰明显代表超过一种样品成份时(例如,有肩膀形的变宽的峰或在两个或多个峰间有谷),选择合适的分析谱和背景谱很重要。
7.6.1.6观察合适离子的萃取离子电流曲线图有助于选取色谱和对化合物的定性鉴别。当分析物共同洗脱时(例如,只有一个色谱峰是明显的),可能会达到此鉴别的标准,但每一分析物会含有共同洗脱化合物的外来离子。
7.6.2对于含有与校正标准物质无关成份的样品,应通过图书馆搜索作试探性识别。是否需要进行此类的鉴别由所进行分析的目的决定。数据系统图书馆的搜索应不要使用正常化的途径,那可能会错误地显示质谱库或当作比较时,显示不能识别此质谱图。
例如《资源保护和开采法》允许或是《废弃物抛弃要求》要求报告非目标分析物,只有通过目视比较样品的谱图同最接近的谱库中的谱图,分析者才可作试探性的鉴别,其指导原则如下:
(1)参考谱图中主要离子的相对强度(离子的相对强度大于大多数富有离子的10%)应在样质量谱图中体现出来。
(2)主要离子的相对强度相差不超过±20%(例如,标准谱中丰度为50%的离子,相应的样品离度必须在30%-70%之间。)
(3)参考谱中存在的分子离子也应在样品谱图中存在。
(4)对于参考谱中不存在但样品谱图中存在的离子应该加以检查,以确定是否有可能背景污染或是存在共同洗脱的化合物。
(5)参考谱中存在但样品谱中不存在的离子,应检查是否由于背景污染或共同洗脱峰的原因而将它从样品谱中减去。数据系统图书馆谱库的减少有时会导致这些差异。
7.7定性分析
7.7.1当一种化合物被鉴别后,化合物的量化是基于萃取离子电流曲线图中的主要特征离子的丰度积分。
7.7.2如果一种化合物响应因子的相对标准差小于或等于15%,则可利用初始校正数据的平均响应因子(RF)求出萃取物中化合物的浓度。参考方法8000第7.0节有关内标校正及线性和非线性校正的公式。
7.7.3当可以实行时,应评估样品中鉴别到的非目标分析物的浓度(第7.6.2节)。使用相同的公式进行计算,公式只作如下修正:A X和A IS来自于总的离子色谱图中,化合物的回应因子值假定为1。
7.7.4报告的浓度结果应显示(1)此结果为估计值;(2)哪种内标物质被用来测试此浓度.使用不含干扰的最近的内标物.
7.7.5多种组成成份的化合物(例如,毒杀芬,氯化联二苯树脂,等等)的量化不在方法8270的范围之内.一般地,量化使用GC/ECD,方法为8081或8082.然而,当浓缩的样品萃取物中化合物的浓度至少为10ng/μL时,方法8270可以用来证明这些化合物的鉴别.
7.7.6同分异构体会产生非常相似的质谱图.当它们的GC保留时间差别足够大时,每种同分异构体应单独量化.如果两种同分异构体产生的质谱峰间的谷的高度小于两峰高度和的25%,则可以达到足够的GC分辨率.另外,同分异构体被当作同分异构对来量化.非对映的同分异构对(例如,异黄樟与异黄樟素)可以通过GC方法分离,并将结果相加作为两种化合物的和的结果报告。
8.0质量控制
8.1相关的质量控制程序请参见本手册第一章及方法8000.质量控制程序确保正确地操作各种样品的配制及/或样品的加入技术.样品的加入程序在方法3500中可以找到.每个实验室都应当维持正式的质量保证项目.实验室也应当保留记录,以便将产生数据的质量文件化.
8.2评估GC系统操作必要的质量控制程序在方法8000第7.0章可以找到.它包括校正验证及样品的色谱分析.另外,仪器的质量控制要求可以在方法8270的以下几节找
安徽建筑大学 现代水分析技术论文 专业:xx级市政工程 学生姓名:xxx 学号:xxx 课题:气相色谱法基本原理及其应用指导教师:xxx xx年xx月xx日
气相色谱法基本原理及其应用 xx (安徽建筑工业学院环境与能源工程学院,合肥,230601) 摘要:气相色谱法是分离混合物中各组分的一种有效的手段,其中气相色谱仪是20世纪50年代末在多数科学家的共同努力下诞生的。本文针对气相色谱法的起源与发展历程、工作原理与特点、在环境水污染物分析领域的应用进行了详细的概述,并列举了饮用水中挥发性有机物的气相色谱检测方法,同时提出了该方法新的发展前景。它的发展已在环境监测、水污染控制领中得到了广泛的应用。 关键词:气相色谱法;发展历程;工作原理;水污染物分析 1.气相色谱法的起源与发展历程 (1)气相色谱法的起源 色谱的发现首先认识到这种分离现象和分离方法大有可为的是俄国的植物学家Tswett。Tswett于1903年在波兰华沙大学研究植物叶子的组成时,将叶绿素的石油醚抽提液倒入装有碳酸钙吸附剂的玻璃管上端,然后用石油醚进行淋洗,结果不同色素按吸附顺序在管内形成一条不同颜色的环带,就像光谱一样。1906年,Tswett在德国植物学杂志上发表的一篇论文中首次把这些彩色环带命名为“色谱图”,玻璃管称为“色谱柱”,碳酸钙称为“固定相”,石油醚称为“流动相”。Tswett开创的方法叫做“液-固色谱法”[1-2],这就是色谱法的起源。 1941年,英国科学家Martin和Synge在研究液-液分配色谱时,预言可以使用气体作流动相,即气-夜色谱法。他们在1941年发表的论文中写到“流动相不一定是液体,也可以是蒸气,如以永久性气体带动挥发性混合物,在色谱柱中通过装有浸透不挥发性溶剂的固体时,可以得到很好的分离”[3]。1950年,Martin和James使用硅藻土助滤剂做载体,硅油为固定相,用气体流动相对脂肪酸进行精细分离,这就是气^液分配色谱的起源。后来,他们在1952年的Biochemical Journal上又连续发表了3篇论文[4-6],叙述了用气相色谱分离低碳数脂肪酸、挥发性胺和吡啶类同系物的方法,这标志着气相色谱法正式进入历史舞台。当时在石油化工的分析中,正当传统的分析方法无能为力时,气相色谱法就像及时雨一样,成为化学分析的得力助手。从此,科学家对气相色谱法的研究逐步展开。 (2)气相色谱法的发展 在历史上,气相色谱法的发展总是和气相色谱仪器的发展密不可分。每一种气相色谱新技术的出现,往往都伴随着气相色谱仪器的改进。因此,了解气相色谱法的发展历史可以从气相色谱仪的发展入手。历史上最早的气相色谱仪1947年由捷克色谱学家Jaroslav Janak发明的。该仪器以C为流动相、杜马测氮管为检测器测定分离开的气体体积。在样品和CA 进入测氮管之前,通过KOH溶液吸收掉CA,按时间记录气体体积的增量。这台仪器虽然简陋,但对当时的气相色谱研究起到了巨大的推动作用。Jaroslav Janak发明的气相色谱仪也有一些明显的不足:它只能测室温下为气体的样品, 样品中的CA不能被测定,而且没有实现自动化。20世纪50年代末,它逐渐被更先进的气相色谱仪所取代。W55年,第一台商品化气相色谱仪诞生,标志着气相色谱仪的发展进入了崭新的时代。 现代气相色谱仪主要由5个系统组成,即气路系统、进样系统、分离系统、温度控制系统与检测记录系统。气路系统与温控系统自气相色谱诞生以来很少有突破性的进展。气路系统主要朝自动化方向发展,20世纪90年代出现了采用电子压力传感器和电子流量控制器,通过计算机实现压力和流量自动控制的电子程序压力流量控制系统,这是气路系统的一大进步[7]。温控系统则基本朝着精细、快速、自动化方向发展。相比之下,进样系统、分离系统与检测记录系统是气相色谱仪的核心组成系统,它们的每一次变革和进步都推动着气相色谱的
归一化法 归一化法有时候也被称为百分法(percent),不需要标准物质帮助来进行定量。它直接通过峰面积或者峰高进行归一化计算从而得到待测组分的含量。其特点是不需要标准物,只需要一次进样即可完成分析。 归一化法兼具内标和外标两种方法的优点,不需要精确控制进样量,也不需要样品的前处理;缺点在于要求样品中所有组分都出峰,并且在检测器的响应程度相同,即各组分的绝对校正因子都相等。归一化法的计算公式如下: 当各个组分的绝对校正因子不同时,可以采用带校正因子的面积归一化法来计算。事实上,很多时候样品中各组分的绝对校正因子并不相同。为了消除检测器对不同组分响应程度的差异,通过用校正因子对不同组分峰面积进行修正后,再进行归一化计算。其计算公式如下: 与面积归一化法的区别在于用绝对校正因子修正了每一个组分的面积,然后再进行归一化。注意,由于分子分母同时都有校正因子,因此这里也可以使用统一标准下的相对校正因子,这些数据很容易从文献得到。 当样品中不出峰的部分的总量X通过其他方法已经被测定时,可以采用部分归一化来测定剩余组分。计算公式如下: 内标法 选择适宜的物质作为预测组分的参比物,定量加到样品中去,依据欲测定组分和参比物在检测器上的响应值(峰面积或峰高)之比和参比物加入量进行定量分析的方法叫内标法。特点是标准物质和未知样品同时进样,一次进样。内标法的优点在于不需要精确控制进样量,由进样量不同造成的误差不会带到结果中。缺陷在于内标物很难寻找,而且分析操作前需要较多的处理过程,操作复杂,并可能带来误差。 一个合适的内标物应该满足以下要求:能够和待测样品互溶;出峰位置不和样品中的组分
重叠;易于做到加入浓度与待测组分浓度接近;谱图上内标物的峰和待测组分的峰接近。内标法的计算公式推导如下: 式中,Ai,As分别为待测组分和内标物的峰面积;Ws,W分别为内标物和样品的质量;Gwi/s是待测组分对于内标物的相对质量校正因子(此值可自行测定,测定要求不高时也可以由文献中待测组分和内标物组分对苯的相对质量校正因子换算求出)。 内加法 在无法找到样品中没有的合适的组分作为内标物时,可以采用内加法;在分析溶液类型的样品时,如果无法找到空白溶剂,也可以采用内加法。内加法也经常被称为标准加入法。 内加法需要除了和内标法一样进行一份添加样品的处理和分析外,还需要对原始样品进行分析,并根据两次分析结果计算得到待测组分含量。和内标法一样,内加法对进样量并不敏感,不同之处在于至少需要两次分析。下面我们用一个实际应用的例子来说明内加法是如何工作的: 题:在分析某混合芳烃样品时,测得样品中苯的面积为1100,甲苯的面积为2000,(其它组分面积略)。精确称取40.00g该样品,加入0.40g甲苯后混合均匀,在同一色谱仪上进混合后样品测到苯的面积为1200,甲苯的面积为2400,试计算甲苯的含量。 分析:本题的分析过程是一个典型的内加法操作,其中内加物为甲苯,待测组分为甲苯和苯。 解:1. 由于进样量并不准确,因此两次分析的谱图很难直接进行对比。为了取得可以对比的一致性,我们通过数字计算调整两次分析苯的峰面积相等。此时由于两次分析苯峰面积相等,因此可以断定两次分析待测样品的进样量是相等的。需要注意的是:此时两次分析的总的进样量并不相等,添加后样品比原始样品调整后的进样量中,多了添加的内标物的量。调整可以用原始样品谱图为依据,也可以用添加后样品谱图为依据。但是通常采用原始样品作为依据以便计算最终结果时比较简单。注意:选用的依据不同,中间计算结果会产生差异,但不会影响最终结果。依据的谱图一旦选定,计算就应该围绕此依据进行。 在以原始样品谱图为依据的情况下,调整添加后样品谱图中甲苯的峰面积如下: 对比两次分析,甲苯的面积增加为2200-2000=200。在两次分析待测样品量相同的情况下,内加物面积的增加来自于内加量。也就是说,由于内加物的加入,导致了内加物峰面积的增
气相色谱仪原理(图文详解) 什么是气相色谱 本章介绍气相色谱的功能和用途,以及色谱仪的基本结构。 气相色谱(GC)是一种把混合物分离成单个组分的实验技术。它被用来对样品组分进行鉴定和定量测定: 基子时间的差别进行分离 和物理分离(比如蒸馏和类似的技术)不同,气相色谱(GC)是基于时间差别的分离技术。 将气化的混合物或气体通过含有某种物质的管,基于管中物质对不同化合物的保留性能不同而得到分离。这样,就是基于时间的差别对化合物进行分离。样品经过检测器以后,被记录的就是色谱图(图1),每一个峰代表最初混合样品中不同的组分。 峰出现的时间称为保留时间,可以用来对每个组分进行定性,而峰的大小(峰高或峰面积)则是组分含量大小的度量。 图1典型色谱图
系统 一个气相色谱系统包括 可控而纯净的载气源.它能将样品带入GC系统进样口,它同时还作为液体样品的气化室色谱柱,实现随时间的分离 检测器,当组分通过时,检测器电信号的输出值改变,从而对组分做出响应 某种数据处理装置图2是对此作出的一个总结。 样品 载气源一^ 进样口一^ 色谱柱一^ 检测器一_ 数据处理」 图2色谱系统 气源 载气必须是纯净的。污染物可能与样品或色谱柱反应,产生假峰进入检测器使基线噪音增大等。推荐使用配备有水分、烃类化合物和氧气捕集阱的高纯载气。见图
钢瓶阀 若使用气体发生器而不是气体钢瓶时,应对每一台GC都装配净化器,并且使气源尽可能靠近仪器的背面。 进样口 进样口就是将挥发后的样品引入载气流。最常用的进样装置是注射进样口和进样阀。注射进样口 用于气体和液体样品进样。常用来加热使液体样品蒸发。用气体或液体注射器穿透隔垫将样品注入载气流。其原理(非实际设计尺寸)如图4所示。
在实际工作中,当我们拿到一个样品,我们该怎样定性和定量,建立一套完整的分析方法是关键,下面介绍一些常规的步骤: 1、样品的来源和预处理方法 GC能直接分析的样品通常是气体或液体,固体样品在分析前应当溶解在适当的溶剂中,而且还要保证样品中不含GC不能分析的组分(如无机盐),可能会损坏色谱柱的组分。这样,我们在接到一个未知样品时,就必须了解的来源,从而估计样品可能含有的组分,以及样品的沸点范围。如果样品体系简单,试样组分可汽化则可直接分析。如果样品中有不能用GC直接分析的组分,或样品浓度太低,就必须进行必要的预处理,如采用吸附、解析、萃取、浓缩、稀释、提纯、衍生化等方法处理样品。 2、确定仪器配置 所谓仪器配置就是用于分析样品的方法采用什么进样装置、什么载气、什么色谱柱以及什么检测器。 一般应首先确定检测器类型。碳氢化合物常选择FID检测器,含电负性基团(F、Cl等)较多且碳氢含量较少的物质易选择ECD检测器;对检测灵敏度要求不高,或含有非碳氢化合物组分时,可选择TCD检测器;对于含硫、磷的样品可选择FPD检测器。 对于液体样品可选择隔膜垫进样方式,气体样品可采用六通阀或吸附热解析进样方法,一般色谱仅配置隔膜垫进样方式,所以气体样品可采用吸附-溶剂解析-隔膜垫进样的方式进行分析。 根据待测组分性质选择适合的色谱柱,一般遵循相似相容规律。分离非极性物质时选择非极性色谱柱,分离极性物质时选择极性色谱柱。色谱柱确定后,根据样本中待测组分的分配系数的差值情况,确定色谱柱工作温度,简单体系采用等温方式,分配系数相差较大的复杂体系采用程序升温方式进行分析。 常用的载气有氢气、氮气、氦气等。氢气、氦气的分子量较小常作为填充柱色谱的载气;氮气的分子量较大,常作为毛细管气相色谱的载气;气相色谱质谱用氦气作为载气。 3、确定初始操作条件 当样品准备好,且仪器配置确定之后,就可开始进行尝试性分离。这时要确定初始分离条件,主要包括进样量、进样口温度、检测器温度、色谱柱温度和载气流速。进样量要根据样品浓度、色谱柱容量和检测器灵敏度来确定。样品浓度不超过10mg/mL时填充柱的进样量通常为1-5uL,而对于毛细管柱,若分流比为50:1时,进样量一般不超过2uL。进样口温度主要由样品的沸点范围决定,还要考虑色谱柱的使用温度。原则上讲,进样口温度高一些有利,一般要接近样品中沸点最高的组分的沸点,但要低于易分解温度。
气相色谱法在环境分析中的应用 摘要:气相色谱法是一种很常见的环境分析检测方法,我们也经常将它应用在水、大气、固废等环境检测中。我们以检测非甲烷烃为例来进行探究和学习,(非甲烷烃是一种对人体健康有害的气体)因此我们利用带有双柱双氢火焰离子化检测器的气相色谱仪(岛津GC2014型)和自己所学的知识来对此进行气相色谱检测。并且通过这次检测来了解和复习流动相、检测器、色谱柱以及温度等色谱条件是如何选择以及定性、定量分析方法。 关键词:非甲烷总烃;气相色谱法;定性、定量分析; 1.非甲烷总烃 非甲烷烃(NMHC通常是指除甲烷以外的所有可挥发的碳氢化合物(其中主要是C2~C8,又称非甲烷总烃。主要包括烷烃、烯烃、芳香烃和含氧烃等组分。大气中的非甲烷总烃超过一定浓度,除直接对人体健康有害外,在一定条件下经日光照射还能产生光化学烟雾,对环境和人类造成危害[1]。 监测环境空气和工业废气中的NMHC有许多方法,但目前多数国家采用气相色谱法。由于直接测定NMHC所用仪器价格昂贵,因此我们采用双柱双氢火焰离子化检测器气相色谱法分别测出总烃和甲烷的含量,两者之差为NMHC的含量。在规定的条件下所测得的NMHC是于气相色谱氢火焰离子化检测器有明显响应的除甲烷外碳氢化合物总量,以碳计[2]。 目前我国基本采用气相色谱法测定非甲烷总烃, 按进样的不同有活性炭吸附一热解吸法及针筒采样一手动进样法,采用活性炭吸附一热解吸法[3]易受到活性炭吸附效率的影响,而针筒采样——手动进样法[4]则重复性较差、易熄火。而我们采用气袋采样—气体自动进样器进样分析气体中非甲烷总烃,而这样也最令人满意。此方法操作简单、重复性好、效率高、干扰少,且可用于其他挥发性有机物,如苯系物等的测定。 2.利用气相色谱法检测非甲烷总烃
气相色谱仪的简介及如何应用 气相色谱仪 气相色谱法适用于分析具有一定蒸气压且热稳定性好的组分,对气体试样和受热易挥发的有机物可直接进行分析,而对500℃以下不易挥发或受热易分解的物质部分可采用衍生化法或裂解法。 一、仪器的组成 气相色谱仪由载气源、进样部分、色谱柱、柱温箱、检测器和数据处理系统组成。进样部分、色谱柱和检测器的温度均在控制状态。 二、对仪器的基本要求 1.对仪器的一般要求 (1)载气源气体氦、氮和氢可用作气相色谱法的流动相,可根据供试品的性质和检测器种类选择载气,除另有规定外,常用载气为氮气。 (2)进样部分进样方式一般可采用溶液直接进样或顶空进样。采用溶液直接进样时,进样口温度应高于柱温30~50℃。顶空进样适用于固体和液体供试品中挥发性组分的分离和测定。 (3)色谱柱根据需要选择。新填充柱和毛细管柱在使用前需老化以除去残留溶剂及低分子量的聚合物,色谱柱如长期未用,使用前应老化处理,使基线稳定。 (4)柱温箱柱温箱温度的波动会影响色谱分析结果的重现性,因此柱温箱控温精度应在±1℃,且温度波动小于每小时0.1℃。 (5)检测器适合气相色谱法的检测器有火焰离子化检测器(FID)、热导检测器(TCD)、氮磷检测器(NPD)、火焰光度检测器(FPD)、电子捕获检测器(ECD)、质谱检测器(MS)等。火焰离子化检测器对碳氢化合物响应良好,适合检测大多数的药物;氮磷检测器对含氮、磷元素的化合物灵敏度高;火焰光度检测器对含磷、硫元素的化合物灵敏度高;电子捕获检测器适于含卤素的化合物;质谱检测器还能给出供试品某个成分相应的结构信息,可用于结构确证。除另有规定外,火焰离子化检测器一般用氢气作为燃气,空气作为助燃气。在使用火焰离子化检测器时,检测器温度一般应高于柱温,并不得低于150℃,以免水汽凝结,通常为250~350℃。 (6)数据处理系统目前多用计算机工作站。 药典规定,各品种项下规定的色谱条件,除载气、检测器、固定液品种及特殊指定的色谱柱材料不得改变外,其余如色谱柱内径、长度、载体牌号、粒度、固定液涂布浓度、载气流速、柱温、进样量、检测器的灵敏度等,均可适当改变,以适应具体品种并符合系统适用性试验
气相色谱法的应用 气相色谱法在石油工业中的应用 ⑴石油气的分析石油气(C1~C4)的成分分析,目前都采用气相色谱法。以25%丁酮酸乙酯为固定液,6201担体,柱长12.15m,内径4mm,柱温12℃,氢为载气,流速25ml/nin,热导池电桥电流120~150mA, C1~C4各组分得较好的分离见图10。图10 石油在丁酮酸乙酯柱上的分离1-空气;2-乙烷;3-乙烯;4-二氧化碳;5-丙烷;6-丙烯;7-异丁烷8-乙炔;9-正丁烷;10-正丁烯;11-异丁烯12- 反丁烯-2,3;13- 顺丁烯-2,4;14-丁二烯北京化工研究院近期研究出用多孔氧化铝微球色谱固定相,对C1~C4烃分离很好,柱长2m,内径2mm,内填充0.3%阿皮松L,改性?-Al2O3,微球120~130目;柱温85℃,氮为载气,流速15ml/min,氢火焰离子化检测器。分离谱见图11. 此外吉林化学工业公司研究院还研制了石墨化炭黑和改性石墨化炭黑色谱固定相分离C1~C4烃。⑵石油馏的的分析气相色谱法分析石油馏分的效能与分析速度是精密分馏等化学方法所不能比拟的。如一根60m长、内径0.17mm的弹性石英毛细管柱,内涂OV-101,在程序升温条件下(柱温40~90℃)进样0.6?1,分流比150:1,分析了65~165℃大港直馏气油。用一根30m长、内径0.25mm 毛细管柱,涂PEG1500,柱温80℃,汽化100℃,氮为载气,分流比100:1,汽油中微量芳香烃得到很好的分离(见图12)。图11 低级烃类的气相色谱分离图1-CH4;2-C2H6;3-C2 H4;4-C3 H8;5-C2 H2;6-C8 H6;7-iC4 H10;8-nC4 H10;9-丙二烯;10-丁烯-1;11-iC5 H12 12--i C4 H6;13- 反丁烯-2;14- 顺丁烯-2;15-丁二烯16-丙炔图12汽微量芳烃的油中色谱分离1-苯;2-甲苯;3-乙苯;4-对二甲苯;5-一间二甲苯; 6-邻二甲苯 气相色谱法在环境科学中的应用 我国在环境科学研究、监督检测中,广泛使用气相色谱法测定大气和水中痕量胡害物质。 ⑴大气中微量-氧化碳的分析 汽车尾气中含有一氧化碳,工业锅炉和家用煤炉燃烧不完全放出一氧化碳,都污染环境。大气中痕量一氧化碳常用转化法没定。国产SP-2307色谱仪具有转化装置,使CO转化为CH4。CO+3H2Ni催化/380℃→CH4+H2O 色谱柱固定相可用5A筛分子,GDX-104,Porpak Q等,以分子筛为例,13X或5A分子筛60~80目(先经500~550℃活化2小时)以氢气载气, 57ml/nin;氢焰检测器;空气400ml/min;尾吹氮气80ml/min。柱长2m,内径2mm,柱温36℃,检测室130℃,转化炉380v;进样量1mm。可测大气中ppm级一氧化碳。
气相色谱法测定丁醇中少量甲醇含量 一、实验目的 1. 掌握用外标法进行色谱定量分析的原理和方法。 2. 了解气相色谱仪氢火焰离子检测器FID的性能和操作方法。 3. 了解气相色谱法在产品质量控制中的应用。 4. 学习气相色谱法测定甲醇含量的分析方法。 二、实验原理 在丁醇生产的过程中,不可避免地有甲醇产生。甲醇是无色透明的具有高度挥发性的液体,是一种对人体有害的物质。甲醇在人体内氧化为甲醛、甲酸,具有很强的毒性,对神经系统尤其是视神经损害严重,人食入 5 g 就会出现严重中毒,超过 12. 5 g 就可能导致死亡,在白酒的发酵过程中,难以将甲醇和乙醇完全分离,因此国家对白酒中甲醇含量做出严格规定。根据国家标准(GB10343-89),食用酒精中甲醇含量应低于0.1g?L-1(优级)或0.6 g?L-1(普通级)。 气相色谱法是一种高效、快速而灵敏的分离分析技术,具有极强的分离效能。一个混合物样品定量引入合适的色谱系统后,样品被气化后,在流动相携带下进入色谱柱,样品中各组分由于各自的性质不同,在柱内与固定相的作用力大小不同,导致在柱内的迁移速度不同,使混合物中的各组分先后离开色谱柱得到分离。分离后的组分进入检测器,检测器将物质的浓度或质量信号转换为电信号输给记录仪或显示器,得到色谱图。利用保留值可定性,利用峰高或峰面积可定量。 外标法是在一定的操作条件下,用纯组分或已知浓度的标准溶液配制一系列不同含量的标准溶液,准确进样,根据色谱图中组分的峰面积(或峰高)对组分含量作标准曲线。在相同操作条件下,依据样品的峰面积(或峰高),从标准曲线上查出其相应含量。利用气相色谱可分离、检测丁醇中的甲醇含量,在相同的操作条件下,
永久性气体色谱分析 1.方法原理 以13X或5A分子筛为固定相,用气固色谱法分析混合气中的氧、氮、甲烷、一氧化碳,用纯物质对照进行定性,再用峰面积归一化法计算各个组分的含量。 2.仪器和试剂 ①仪器气相色谱仪,备有热导池检测器;皂膜流量计;秒表。 ②试剂13X或5A分子筛(60~80目);使用前预先在高温炉内,于350℃活化4h后备用。纯氧气、氮气、甲烷、一氧化碳装入球胆或聚乙烯取样袋中。氢气装在高压钢瓶内。3.色谱分析条件 固定相:13X或5A分子筛(60~80目);不锈钢填充柱管φ4mm×2m;柱温:室温。 载气:氢气,流量30mL/min 检测器:热导池检测器,桥流200mA;衰减1/2~1/8,检测室温度:室温。 气化室:室温,进样量用六通阀进样,定量管0.5mL。 4.定性分析 记录各组分从色谱柱流出的保留时间,用纯物质进行对照。 5.定量分析 由谱图中测得各个组分的峰高和半峰宽计算各组分的峰面积。已知氧、氮、甲烷、一氧化碳的相对摩尔校正因子分别为2.50、2.38、2.80、2.38。再用峰面积归一法就可计算出各个组分的体积百分数(%)。
白酒中主要成分的色谱分析 1.方法原理 白酒的主要成分为醇、酯和羟基化合物,由于所含组分较多,且沸点范围较宽,适合用程序升温气相色谱法进行分离,并用氢火焰离子化检测器进行检测。 为分离白酒中的主要成分可使用填充柱或毛细管柱,常用的填充柱固定相为GDX-102;16%邻苯二甲酸二壬酯+7%吐温-60/硅烷化101白色载体(60~80目);10%聚乙二醇20M/有机载体402(80~100目);15%吐温-60+15%司班-60/6201红色载体(60~80目)等。也可使用以聚乙二醇20M或FFAP交联制备的石英弹性毛细管柱。 2.仪器和试剂 ①仪器带有分流进样器和氢火焰离子化检测器的气相色谱仪、皂膜流量计、微处理机。 ②试剂氮气、氢气、压缩空气,与白酒中主要成分对应的醛、醇、酯的色谱纯标样。 3.色谱分析条件 色谱柱:冠醚+FFAP交联石英弹性毛细管柱φ0.25mm×30m,固定液液膜厚度df=0.5um。程序升温:50℃(6min)以40℃/min升温至220℃(1min)。 载气:氮气,流量1mL/min。燃气:氢气,流量50mL/min。助燃气:压缩空气,流量500mL/min。 检测器:氢火焰离子化检测器,高阻1010Ω,衰减1/4~1/16,检测室温度200℃。 气化室:250℃,分流进样分流比1:100,进样量0.2uL。 4.定性分析 记录各组分的保留时间和保留温度,用标准样品对照。 5.定量分析 以乙酸正丁酯作内标,用内标法定量。
白酒气相色谱分析方法 白酒香味成份复杂,除乙醇和水外,还有大量芳香组分存在。构成白酒质量风格的是酒内所含的香味成分的种类以及其量比关系。应用气相色谱法能快速而准确地测出白酒中的醇类、酯类、有机酸类、碳基化合物、酚类化合物以及高沸点化合物等成分的含量。 一、填充柱DNP柱测定白酒中醇、酯等组分(一般酒厂需要,白酒) (一)DNP柱直接进样法测定白酒中主要醇、酯成份 白酒中醇和酯是主要香味成份。吸取原样品进行色谱分析,其优点是:操作简便,测定结果准确性高、快速;缺点是:极其微量的组分不易检出。 1样品的配制 ●2%内标的配制: 吸取2mL的内标--乙酸正丁酯于1OOmL的容量瓶中,(因内标物易挥发,可在瓶内先放少量酒精),用55%-60%的乙醇定容。 ●1-2%标样的配制: 分别吸取乙醛、甲醇、正丙醇、仲丁醇、乙缩醛、正丁醇、异戊醇、(正己醇)、(糠醛)各lmL,乙酸乙酯、丁酸乙酯、戊酸乙酯、乳酸乙酯、己酸乙酯、乙
酸异戊酯)各2mL一起加入1OOmL容量瓶中,用55%-60%(V/V)的乙醇定容,混匀后组成标样。(在容量瓶中先加少许乙醇,以防挥发) ●混标的配制: 分别用移液管吸取标样lOmL和内标5mL,用55%-60%(V/V)的乙醇定容到1OOmL,混匀后(可分装)待用。 混标中各组分i及内标含量计算公式: mi=ci×Vi×di×lO00 ms=cs×Vs×ds×lO00 式中:mi/ms—混标中各组分i/内标的含量(mg/l0OmL); ci/cs—混标中各组分i/内标的浓度(V/V) Vi/Vs—混标中各组分i/内标的体积(mL) ; di/ds—混标中各组分i/内标的密度(g/mL) ; 1000—算成以mg为单位的系数。 例:计算混标中正丁醇的含量 m正丁醇=1%×lOml×0.809g/ml×lO00=80.9mg/100ml混标样
-科苑论谈 气相色谱法在分析中的应用 王颖石 (黑化集团有限公司,黑龙江齐齐哈尔161041) 摘要:简述气相色谱法近年来的发展及在分析中所起到的重要作用,详细阐述气相色谱法的工作原理、方法特点、操作流程及气相色谱曲线的特点。 关键词:气相色谱;色谱柱;色谱峰;载气 前言:气相色谱法是近五十年来迅速发展起来的一种新型分离,分析技术,在石油炼制、基本有机原料、高分子、医药、原子能、冶金工业中得到了广泛的应用。对保证工业生产的正常进行和提高产品质量起到了重要的作用。在许多生产部门,气相色谱分析法逐步代替了化学分析法。当前随着我国石油化学工业的迅速发展,气相色谱法在石油、化工生产中已成为中间控制分析中的一种不可缺少的分析方法了。 近年来电子计算机和专用的微型电子计算机已和气相色谱仪联用,可自动对分析结果进行数据处理,对于提高分析速度、改善分析结果的准确性及实现生产过程高自动化起到了重要的作用。现就气相色谱法的原理、特点及流程作以详细阐述。 1气相色谱法工作原理
气相色谱的工作原理是利用试样中各组份在色谱柱中的气相和固定液相间的分配系数不同,当汽化后的试样被载体带入色谱中运行时,组份就在其中的两相间进行反复多次的分配(吸附-脱附或溶解-放出),由于固定相对各组份的吸附或溶解能力不同,(即保留作用不同),各组份在色谱柱中的运行速度也就不同,经过一定柱长后,便彼此分离,按顺序离开色谱柱,进入检测器,产生的离子流经讯号放大后,在记录仪上就描绘各组份的曲线图,称为色谱峰。根据色谱峰的峰高或峰面积就可定量测定出样品中各级份的含量。 2气相色谱法的主要特点 气相色谱法在应用中的主要特点是选择性高、分离效率高、灵敏度高、分析速度快。 2.1选择性高 选择性高是指气相色谱法对性质极为接近的物质,具有很强的分离能力。如在石油化工生产中比较难解决的碳四烯烃异构体的分离;原子能工业中氢的三种同位素:氢、氘、氚的分离;医药和生物化学中结构复杂的旋光异构体的分离。现都可采用气相色谱法来解决。 2.2分离效率高 分离效率高是指气相色谱法能分离分配系数很接近的组份一根1~2m的色谱柱,柱效率可达几千块理论塔板数,因而对组成复杂的或难以分离的物质,经过色谱柱进行反复多次的分配平衡(或吸附平衡),最终均可达到分离的目的。 2.3灵敏度高
气相色谱仪原理、结构及操作 1、基本原理 气相色谱(GC )是一种分离技术。实际工作中要分析的样品往往是复杂基体中的多组分混合物,对含有未知组分的样品,首先必须将其分离,然后才能对有关组分进行进一步的分析。混合物的分离是基于组分的物理化学性质的差异,GC 主要是利用物质的沸点、极性及吸附性质的差异来实现混合物的分离。待分析样品在汽化室汽化后被惰性气体(即载气,一般是N2、He 等)带入色谱柱,柱内含有液体或固体固定相,由于样品中各组分的沸点、极性或吸附性能不同,每种组分都倾向于在流动相和固定相之间形成分配或吸附平衡。但由于载气是流动的,这种平衡实际上很难建立起来,也正是由于载气的流动,使样品组分在运动中进行反复多次的分配或吸附/解附,结果在载气中分配浓度大的组分先流出色谱柱,而在固定相中分配浓度大的组分后流出。当组分流出色谱柱后,立即进入检测器,检测器能够将样品组分的存在与否转变为电信号,而电信号的大小与被测组分的量或浓度成比例,当将这些信号放大并记录下来时,就是如图2所示的色谱图(假设样品分离出三个组分),它包含了色谱的全部原始信息。在没有组分流出时,色谱图的记录是检测器的本底信号,即色谱图的基线。 2、气相色谱结构及维护 2.1 进样隔垫 进样隔垫一般为硅橡胶材料制成,一般可分普通型、优质型和高温型三种,普通型为米黄色,不耐高温,一般在200℃以下使用;优质型可耐温到300℃;高温型为绿色,使用温度可高于350℃,至色谱柱最高使用温度的400℃。正因为进样隔垫多为硅橡胶材料制成,其中不可避免地含有一些残留溶剂和/或低分子齐聚物,另外由于汽化室高温的影响,硅橡胶会发生部分降解,这些残留的溶剂和降解产物如果进入色谱柱,就可能出现“鬼峰”(即不是样品本身的峰),从而影响分析。解决的办法有:一是进行“隔垫吹扫”,二是更换进样隔垫。一般更换进样隔
气相色谱法(附答案) 一、填空题1. 气相色谱柱的老化温度要高于分析时最高柱温_____℃,并低于固定液的最高使用温度,老化时,色谱柱要与_____断开。答案:5~10 检测器 2. 气相色谱法分离过程中,一般情况下,沸点差别越小、极性越相近的组分其保留值的差别就_____,而保留值差别最小的一对组分就是_____物质对。答案:越小难分离 3.气相色谱法分析非极性组分时应首先选用_____固定液,组分基本按沸点顺序出峰,如烃和非烃混合物,同沸点的组分中_____大的组分先流出色谱柱。答案:非极性极性 4.气相色谱法所测组分和固定液分子间的氢键力实际上也是一种_____力,氢键力在气液色谱中占有_____地位。答案:定向重要5.气相色谱法分离中等极性组分首先选用_____固定液,组分基本按沸点顺序流出色谱柱。答案:中极性 6.气相色谱分析用归一化法定量的条件是______都要流出色谱柱,且在所用检测器上都能_____。 答案:样品中所有组分产生信号 7.气相色谱分析内标法定量要选择一个适宜的__,并要求它与其他组分能__。答案:内标物完全分离 8.气相色谱法常用的浓度型检测器有_____和_____。答案:热导检
测器(TCD) 电子捕获检测器(ECD) 9. 气相色谱法常用的质量型检测器有_____和_____。答案:氢火焰检测器(FID) 火焰光度检测器(FPD) 10. 电子捕获检测器常用的放射源是_____和_____。答案:63Ni 3H 11. 气相色谱分析中,纯载气通过检测器时,输出信号的不稳定程度称为_____。答案:噪音 12. 顶空气体分析法是依据___原理,通过分析气体样来测定__中组分的方法。答案:相平衡平衡液相 13. 毛细管色谱进样技术主要有_____和______。答案:分流进样不分流进样 14. 液—液萃取易溶于水的有机物时,可用______法。即用添加_____来减小水的活度,从而降低有机化合物的溶解度。答案:盐析盐15.气相色谱载体大致可分为______和______。答案:无机载体有机聚合物载体 16.所谓气相色谱固定液热稳定性好,主要是指固定液在高温下不发生__、__和分解。答案:聚合交联 17. 气相色谱程序升温的方式有_____升温和_____升温。答案:线性非线性 18.气相色谱法分析中,不同的色谱柱温会对柱效、_____、_____、_____和产生影响。
色谱法的分类及其原理 (一)按两相状态 气相色谱法:1、气固色谱法2、气液色谱法 液相色谱法:1、液固色谱法2、液液色谱法 (二)按固定相的几何形式 1、柱色谱法(column chromatography):柱色谱法是将固定相装在一 金属或玻璃柱中或是将固定相附着在毛细管内壁上做成色谱柱,试样从柱 头到柱尾沿一个方向移动而进行分离的色谱法 2、纸色谱法(paper chromatography ):纸色谱法是利用滤纸作固定液的载体,把试样点在滤纸上,然后用溶剂展开,各组分在滤纸的不同位置 以斑点形式显现,根据滤纸上斑点位置及大小进行定性和定量分析。 3、薄层色谱法(thin-layer chromatography, TLC):薄层色谱法是将适 当粒度的吸附剂作为固定相涂布在平板上形成薄层,然后用与纸色谱法类 似的方法操作以达到分离目的。 (三)按分离原理 按色谱法分离所依据的物理或物理化学性质的不同,又可将其分为: 1、吸附色谱法:利用吸附剂表面对不同组分物理吸附性能的差别而使之分离的色谱法称为吸附色谱法。适于分离不同种类的化合物(例如,分离醇类与芳香烃)。
2、分配色谱法:利用固定液对不同组分分配性能的差别而使之分离的色 谱法称为分配色谱法。 3、离子交换色谱法:利用离子交换原理和液相色谱技术的结合来测定溶液中阳离子和阴离子的一种分离分析方法,利用被分离组分与固定相之间发生离子交换的能力差异来实现分离。离子交换色谱主要是用来分离离子或可离解的化合物。它不仅广泛地应用于无机离子的分离,而且广泛地应用于有机和生物物质,如氨基酸、核酸、蛋白质等的分离。 4、尺寸排阻色谱法:是按分子大小顺序进行分离的一种色谱方法,体积大的分子不能渗透到凝胶孔穴中去而被排阻,较早的淋洗出来;中等体积的分子部分渗透;小分子可完全渗透入内,最后洗出色谱柱。这样,样品分子基本按其分子大小先后排阻,从柱中流出。被广泛应用于大分子分 级,即用来分析大分子物质相对分子质量的分布。 5、亲和色谱法:相互间具有高度特异亲和性的二种物质之一作为固定相,利用与固定相不同程度的亲和性,使成分与杂质分离的色谱法。例如利用酶与基质(或抑制剂)、抗原与抗体,激素与受体、外源凝集素与多糖类及核酸的碱基对等之间的专一的相互作用,使相互作用物质之一方与不溶性担体形成共价结合化合物,用来作为层析用固定相,将另一方从复杂的混合物中选择可逆地截获,达到纯化的目的。可用于分离活体高分子物质、过滤性病毒及细胞。或用于对特异的相互作用进行研究。 (四)按原理 色谱过程的本质是待分离物质分子在固定相和流动相之间分配平衡的过程,不同的物质在两相之间的分配会不同,这使其随流动相运动速度各不相同,随着流动相的运动,混合物中的不同组分在固定相上相互分离。
气相色谱分析方法的建立
内标法与外标法 一、内标法 什么叫内标法?怎样选择内标物? 内标法是一种间接或相对的校准方法。在分析测定样品中某组分含量时,加入一种内标物质以校谁和消除出于操作条件的波动而对分析结果产生的影响,以提高分析结果的准确度。 内标法在气相色谱定量分析中是一种重要的技术。使用内标法时,在样品中加入一定量的标准物质,它可被色谱拄所分离,又不受试样中其它组分峰的干扰,只要测定内标物和待测组分的峰面积与相对响应值,即可求出待测组分在样品中的百分含量。采用内标法定量时,内标物的选择是一项十分重要的工作。理想地说,内标物应当是一个能得到纯样的己知化合物,这样它能以准确、已知的量加到样品中去,它应当和被分析的样品组分有基本相同或尽可能一致的物理化学性质(如化学结构、极性、挥发度及在溶剂中的溶解度等)、色谱行为和响应特征,最好是被分析物质的一个同系物。当然,在色谱分析条什下,内标物必须能与样品中各组分充分分离。需要指出的是,在少数情况下,分析人员可能比较关心化台物在一个复杂过程中所得到的回收率,此时,他可以使用一种在这种过程中很容易被完全回收的化台物作内标,来测定感兴趣化合物的百分回收率,而不必遵循以上所说的选择原则。 在使用内标法定量时,有哪些因素会影响内标和被测组分的峰高或峰面积的比值? 影响内标和被测组分峰高或峰面积比值的因素主要有化学方面的、色谱方面的和仪器方面的三类。 由化学方面的原因产生的面积比的变化常常在分析重复样品时出现。 化学方面的因素包括: 1、内标物在样品里混合不好; 2、内标物和样品组分之间发生反应, 3、内标物纯度可变等。 对于一个比较成熟的方法来说,色谱方面的问题发生的可能性更大一些,色谱上常见的一些问题(如渗漏)对绝对面积的影响比较大,对面积比的影响则要小一些,但如果绝对面积的变化已大到足以使面积比发生显著变化的程度,那么一定有某个重要的色谱问题存在,比如进样量改变太大,样品组分浓度和内标浓度之间有很大的差别,检测器非线性等。进样量应足够小并保持不变,这样
气相色谱仪用途和分析 一、气相色谱仪用途和应用领域主要有以下方面: 石油和石油化工分析:油气田勘探中的化学分析、原油分析、炼厂气分析、模拟蒸馏、油料分析、单质烃分析、含硫/含氮/含氧化合物分析、汽油添加剂分析、脂肪烃分析、芳烃分析。 环境分析:大气污染物分析、水分析、土壤分析、固体废弃物分析。 食品分析:农药残留分析、香精香料分析、添加剂分析、脂肪酸甲酯分析、食品包装材料分析。 药物和临床分析:雌三醇分析、儿茶酚胺代谢产物分析、尿中孕二醇和孕三醇分析、血浆中睾丸激素分析、血液中乙醇/麻醉剂及氨基酸衍生物分析。 农药残留物分析:有机氯农药残留分析、有机磷农药残留分析、杀虫剂残留分析、除草剂残留分析等。 精细化工分析:添加剂分析、催化剂分析、原材料分析、产品质量控制。 聚合物分析:单体分析、添加剂分析、共聚物组成分析、聚合物结构表征/聚合物中的杂质分析、热稳定性研究。 合成工业:方法研究、质量监控、过程分析。 二、分析实例: (一)天然气常量分析: 选用热导检测器,适用于城市燃气用天然气O2、N2、CH4、CO2、C2H6、C3H8、i-C40、n-C40、i-C50、n-C50等组分的常量分析。分析结果符合国标GB10410.2-89。 (二)人工煤气分析: 选用热导检测器、双阀多柱系统,自动或手动进样,适用于人工煤气中H2、O2、N2、CO2、CH4、C2H4、C2H6、C3H6等主要成分的测定。分析结果符合国标GB10410.1-89。 (三)液化石油气分析①: 选用热导检测器、填充柱系统、阀自动或手动切换,并配有反吹系统,适用于炼油厂生产的液化石油气中C2-C4及总C5烃类组成的分析(不包括双烯烃和炔烃)。分析结果符合SH/T10230-92。 液化石油气分析②:
实验一气相色谱法分析苯系物 一、实验目的: 1.掌握气相色谱法的基本原理和定性、定量方法。 2.学习纯物质对照法定性和归一化法定量的分析方法。 3.了解气相色谱的仪器组成、工作原理以及数据采集、数据分析的基本操作。 二、实验原理: 气相色谱方法是利用试样中各组份在气相和固定液相间的分配系数不同将混合物分离、测定的仪器分析方法,特别适用于分析含量少的气体和易挥发的液体。当汽化后的试样被载气带入色谱柱中运行时,组份就在其中的两相间进行反复多次分配,由于固定相对各组份的吸附或溶解能力不同,因此各组份在色谱柱中的运行速度就不同,经过一定的柱长后,便彼此分离,按流出顺序离开色谱柱进入检测器,被检测,在记录器上绘制出各组份的色谱峰——流出曲线。在色谱条件一定时,任何一种物质都有确定的保留参数,如保留时间、保留体积及相对保留值等。因此,在相同的色谱操作条件下,通过比较已知纯物质和未知物的保留参数或在固定相上的位置,即可确定未知物为何种物质。测量峰高或峰面积,采用外标法、内标法或归一化法,可确定待测组分的质量分数。 1.典型气相色谱仪由以下五大系统组成: A. 载气系统:包括气源、净化干燥管和载气流速控制; 常用的载气有:氢气、氮气、氦气; 净化干燥管:去除载气中的水、有机物等杂质(依次通过分子筛、活性炭等); 载气流速控制:压力表、流量计、针形稳压阀,控制载气流速恒定。 B. 进样装置:进样器+气化室; 气体进样器(六通阀):推拉式和旋转式两种。 试样首先充满定量管,切入后,载气携带定量管中的试样气体进入分离柱; 液体进样器:不同规格的专用注射器,填充柱色谱常用10μL;毛细管色谱常用1μL; 气化室:将液体试样瞬间气化的装置。 C. 色谱柱(分离柱):色谱仪的核心部件。分为填充柱和毛细管柱。 D. 检测系统:色谱仪的眼睛,常用的检测器:热导检测器、氢火焰离子化检测器; E. 温度控制系统:温度是色谱分离条件的重要选择参数; 气化室、分离室、检测器三部分在色谱仪操作时均需控制温度; 气化室:保证液体试样瞬间气化; 分离室:准确控制分离需要的温度。当试样复杂时,分离室温度需要按一定程序控制温度变化,各组分在最佳温度下分离; 检测器:保证被分离后的组分通过时不在此冷凝。
气相色谱法在环境监测中的应用 应用化学02 冷方方200941602038 摘要:气相色谱法是现代分析的主要手段之一。近年来,气相色谱的各个领 域都取得长足的进步和发展。本文介绍了气相色谱法在大气、室内气体、各种水体和其他类型污染物的应用,并阐述了气相色谱的发展趋势。 关键字:气相色谱法,联用技术,环境监测 1前言 色谱法是一种重要的分离分析方法,它是利用不同物质在两相中具有不同的分配系数(或吸附系数、渗透性),当两相作相对运动时,这些物质在两相中进行多次反复分配而实现分离。在色谱技术中,流动相为气体的叫气相色谱,流动相为液体的叫液相色谱。 气相色谱法由于其具有分离效能高、分析速度快、选择性好等优点而被广泛应用于环境样品中的污染物分析、药品质量检验、天然产物成分分析、食品中农药残留量测定、工业产品质量监控等领域。 2气相色谱法现状 气相色谱法广泛用于纯物质中的杂质、环境污染物、食品中有害成分、药物有效成分、代谢物、刑事法医鉴定、石油化工生产中痕量物质等的分析。随着有毒有害有机污染物对空气、水、土壤及粮食、蔬菜的污染日益严重,有机污染物的监测已得到世界各国的重视。常用的CODCr和CODMn的监测方法不能检测出多环芳烃、苯系物、PCB等强致癌物的状况。GC,GC-MS,HPLC法是有机污染物监测的常用方法。尤其是GC法以其相对价格低廉,操作简便,易于推广利用而备受关注。目前,美国、日本和我国在有机污染物监测的方法中,GC法占了80%。 气相色谱分析法在环境水和废水分析中有着广泛的应用,特别是对水中复杂、痕量、多组分有机物分析,GC是强有力的成分分析工具,而MS是能给出最充分信息的结构分析器。二者的结合常常成为首选的分析方法。据报道少数发达国家已将GC/MS系统列为水中有机物的监测分析方法和标准分析方法,成为有力的鉴定工具。 全球性的多环芳烃污染一直为人们关注。多环芳烃主要产生于煤的加工转化工艺中,后随工业排放水进入环境。由于它具有生物诱变性和致癌性,深受各国的关注。复旦大学的陈正夫、陈思华介绍了利用色谱保留值结合质谱信息鉴定多环芳烃在焦化废水形态分布分析中的应用研究。将多环芳烃的Lee保留指数推广到环境监测中的应用条件和范围,探讨全过程跟踪式的焦化废水采样方式,分析方法切实、有效。