
Qualimap:evaluating next generation sequencing alignment data
Fernando Garc′?a-Alcalde 1,2,Konstantin Okonechnikov 2,Jos′e Carbonell 1,Luis Miguel Ruiz 1,Stefan G¨otz 1,Sonia Tarazona 1,Thomas F .Meyer 2and Ana Conesa 1?
1Bioinformatics and Genomics Department,Centro de Investigaci′on Pr′?ncipe Felipe,Valencia,Spain.
2Department of Molecular Biology,Max-Planck Institute for Infection Biology,Berlin,Germany.ABSTRACT Motivation:The sequence alignment/map (SAM)and the binary alignment/map (BAM)formats have become the standard method of representation of nucleotide sequence alignments for next-generation sequencing data.SAM/BAM ?les usually contain information from tens to hundreds of millions of reads.Often,the sequencing technology,protocol,and/or the selected mapping algorithm introduce some unwanted biases in these data.The systematic detection of such biases is a non-trivial task that is crucial to to drive appropriate downstream analyses.Results:We have developed Qualimap,a Java application that
supports user-friendly quality control of mapping data,by considering
sequence features and their genomic properties.Qualimap takes
sequence alignment data and provides graphical and statistical analyses for the evaluation of data.Such quality-control data are vital for highlighting problems in the sequencing and/or mapping processes,which must be addressed prior to further analyses.Availability:Qualimap is freely available from https://www.doczj.com/doc/0a4367231.html, Contact:aconesa@cipf.es 1INTRODUCTION With the advent of next-generation sequencing (NGS),a number of novel alignment methods have been developed (see Trapnell and Salzberg (2009)for a review).The sequence alignment/map (SAM)and the binary alignment/map (BAM)formats have become the standards used for representation of nucleotide sequence alignments for these algorithms (Li et al.,2009).The results from such alignments can be used in subsequent analyses,such as genome-wide comparative studies,to drive conclusions concerning a variety of biological processes,such as gene expression and epigenomic modi?cations,etc.SAM/BAM ?les usually contain the information from tens to hundreds of millions of reads,and the quantity of data contained in these ?les is continuously increasing.Unfortunately,SAM/BAM data ?les frequently contain biases that are introduced by sequencing technologies,during sample preparation,(Harismendy et al.,2009;Metzker,2009)and/or the
?to whom correspondence should be addressed selected mapping algorithm (Flicek and Birney,2009).Therefore,one of the fundamental requirements during analysis of these data is to perform quality control,i.e.to get an idea of how reliable mapping data are,and how well data ?t with the expected outcome.To these ends,we have developed Qualimap,a Java application that aims to facilitate the quality control analysis of mapping data.Recently,some efforts have been made to facilitate this task,for example see SAMStat (Lassmann et al.,2011),RNA-SeQC (DeLuca et al.,2012),or Picard;Qualimap advances this area by providing some additional features (Supplementary Table S1).2THE QUALIMAP APPLICATION Qualimap is a multi-threaded application built in Java and R that provides a Graphical User Interface (GUI)to perform the quality control of alignment sequencing data 1.The ?rst step in the program is to select the type of analysis to be run,which can be:BAM QC for alignment data and -optionally -a set of regions of interest;or Count QC for count data.
When dealing with alignment data,the main input for Qualimap is the
BAM ?le to be analyzed.The application processes information by splitting
the reference genome into a given number of windows (400by default),
collecting the information and parallelizing the process where appropriate.Results are summarized in a dedicated panel and graphically represented in different charts,which can be exported for further analysis.The user can also concentrate the analysis to speci?c regions of interest by including a General Feature Format (GFF)or a Browser Extensible Data (BED)?le.
In this case,the information is shown separately for reads that are mapped inside or outside of the de?ned regions.
Qualimap also provides insights into mapping performance by studying the read counts overlaps with genomic features of interest.These read counts can be loaded into the program as a text ?le that can be directly computed by using a dedicated tool in Qualimap.For example,the saturation rate for the detected features can be analyzed with respect to the sequencing depth,unveiling whether more features could be detected by increasing the sequencing depth.This is of particular interest,for example,in RNA-seq assays.Likewise,it is possible to analyze the read counts separately in user-de?ned groups of features.
1A command-line interface has been also implemented so Qualimap can be incorporated in particular analysis pipelines.
1? The Author (2012). Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@https://www.doczj.com/doc/0a4367231.html, Associate Editor: Prof. Martin Bishop
Bioinformatics Advance Access published August 22, 2012 at Library on July 24, 2013
https://www.doczj.com/doc/0a4367231.html,/Downloaded from
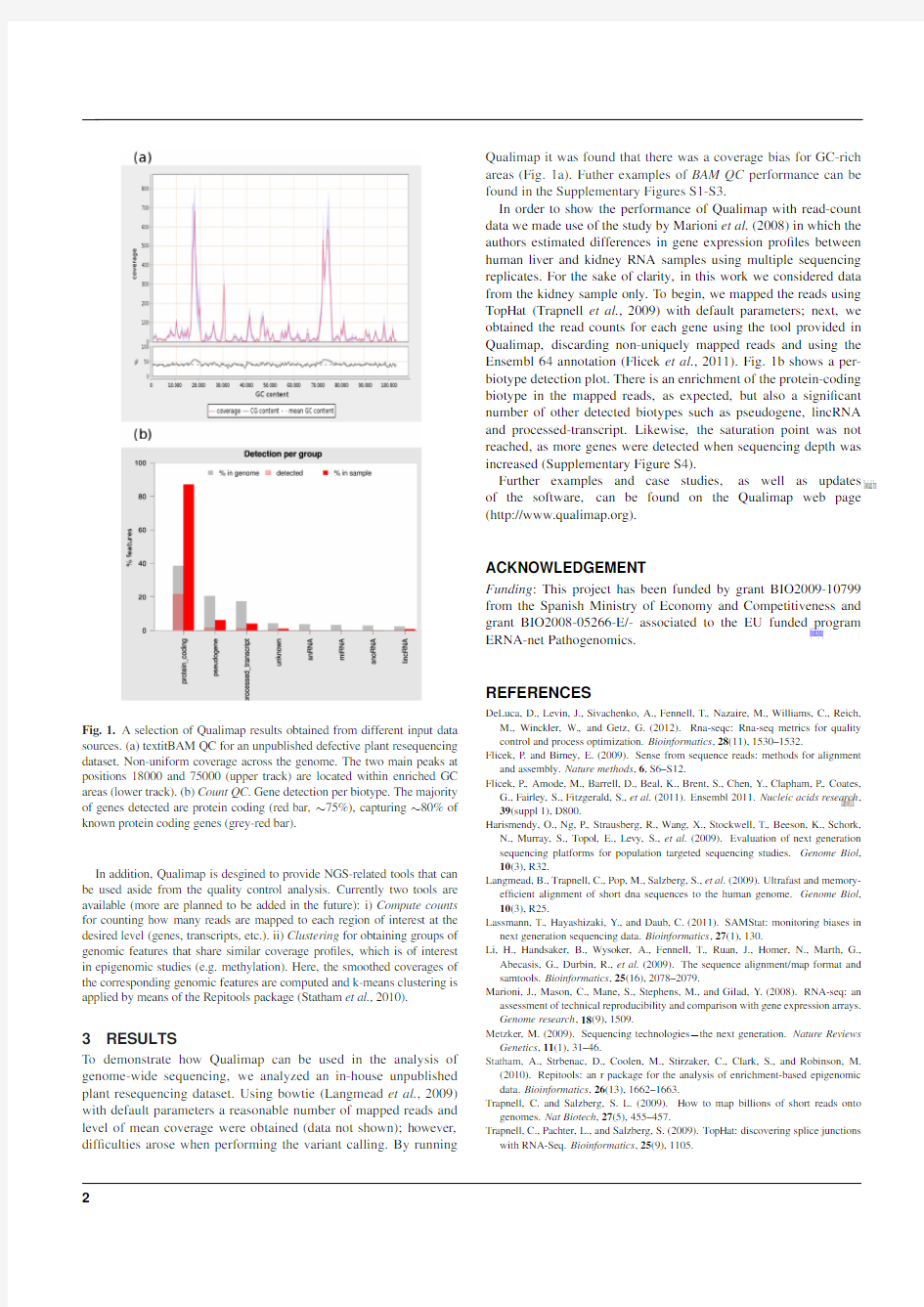
Fig.1.A selection of Qualimap results obtained from different input data sources.(a)textitBAM QC for an unpublished defective plant resequencing dataset.Non-uniform coverage across the genome.The two main peaks at positions18000and75000(upper track)are located within enriched GC areas(lower track).(b)Count QC.Gene detection per biotype.The majority of genes detected are protein coding(red bar,~75%),capturing~80%of known protein coding genes(grey-red bar).
In addition,Qualimap is desgined to provide NGS-related tools that can be used aside from the quality control analysis.Currently two tools are available(more are planned to be added in the future):i)Compute counts for counting how many reads are mapped to each region of interest at the desired level(genes,transcripts,etc.).ii)Clustering for obtaining groups of genomic features that share similar coverage pro?les,which is of interest in epigenomic studies(e.g.methylation).Here,the smoothed coverages of the corresponding genomic features are computed and k-means clustering is applied by means of the Repitools package(Statham et al.,2010).
3RESULTS
To demonstrate how Qualimap can be used in the analysis of genome-wide sequencing,we analyzed an in-house unpublished plant resequencing https://www.doczj.com/doc/0a4367231.html,ing bowtie(Langmead et al.,2009) with default parameters a reasonable number of mapped reads and level of mean coverage were obtained(data not shown);however, dif?culties arose when performing the variant calling.By running Qualimap it was found that there was a coverage bias for GC-rich areas(Fig.1a).Futher examples of BAM QC performance can be found in the Supplementary Figures S1-S3.
In order to show the performance of Qualimap with read-count data we made use of the study by Marioni et al.(2008)in which the authors estimated differences in gene expression pro?les between human liver and kidney RNA samples using multiple sequencing replicates.For the sake of clarity,in this work we considered data from the kidney sample only.To begin,we mapped the reads using TopHat(Trapnell et al.,2009)with default parameters;next,we obtained the read counts for each gene using the tool provided in Qualimap,discarding non-uniquely mapped reads and using the Ensembl64annotation(Flicek et al.,2011).Fig.1b shows a per-biotype detection plot.There is an enrichment of the protein-coding biotype in the mapped reads,as expected,but also a signi?cant number of other detected biotypes such as pseudogene,lincRNA and processed-transcript.Likewise,the saturation point was not reached,as more genes were detected when sequencing depth was increased(Supplementary Figure S4).
Further examples and case studies,as well as updates of the software,can be found on the Qualimap web page (https://www.doczj.com/doc/0a4367231.html,).
ACKNOWLEDGEMENT
Funding:This project has been funded by grant BIO2009-10799 from the Spanish Ministry of Economy and Competitiveness and grant BIO2008-05266-E/-associated to the EU funded program ERNA-net Pathogenomics.
REFERENCES
DeLuca,D.,Levin,J.,Sivachenko,A.,Fennell,T.,Nazaire,M.,Williams,C.,Reich, M.,Winckler,W.,and Getz,G.(2012).Rna-seqc:Rna-seq metrics for quality control and process optimization.Bioinformatics,28(11),1530–1532.
Flicek,P.and Birney,E.(2009).Sense from sequence reads:methods for alignment and assembly.Nature methods,6,S6–S12.
Flicek,P.,Amode,M.,Barrell,D.,Beal,K.,Brent,S.,Chen,Y.,Clapham,P.,Coates,
G.,Fairley,S.,Fitzgerald,S.,et al.(2011).Ensembl2011.Nucleic acids research,
39(suppl1),D800.
Harismendy,O.,Ng,P.,Strausberg,R.,Wang,X.,Stockwell,T.,Beeson,K.,Schork, N.,Murray,S.,Topol,E.,Levy,S.,et al.(2009).Evaluation of next generation sequencing platforms for population targeted sequencing studies.Genome Biol, 10(3),R32.
Langmead,B.,Trapnell,C.,Pop,M.,Salzberg,S.,et al.(2009).Ultrafast and memory-ef?cient alignment of short dna sequences to the human genome.Genome Biol, 10(3),R25.
Lassmann,T.,Hayashizaki,Y.,and Daub,C.(2011).SAMStat:monitoring biases in next generation sequencing data.Bioinformatics,27(1),130.
Li,H.,Handsaker,B.,Wysoker,A.,Fennell,T.,Ruan,J.,Homer,N.,Marth,G., Abecasis,G.,Durbin,R.,et al.(2009).The sequence alignment/map format and samtools.Bioinformatics,25(16),2078–2079.
Marioni,J.,Mason,C.,Mane,S.,Stephens,M.,and Gilad,Y.(2008).RNA-seq:an assessment of technical reproducibility and comparison with gene expression arrays.
Genome research,18(9),1509.
Metzk er,M.(2009).Sequencing technologies?the next generation.Nature Reviews Genetics,11(1),31–46.
Statham,A.,Strbenac,D.,Coolen,M.,Stirzaker,C.,Clark,S.,and Robinson,M.
(2010).Repitools:an r package for the analysis of enrichment-based epigenomic data.Bioinformatics,26(13),1662–1663.
T rapnell,C.and Salzberg,S.L.(2009).How to map billions of short reads onto genomes.Nat Biotech,27(5),455–457.
T rapnell,C.,Pachter,L.,and Salzberg,S.(2009).TopHat:discovering splice junctions with RNA-Seq.Bioinformatics,25(9),1105.
2 at Library on July 24, 201
3 https://www.doczj.com/doc/0a4367231.html,/Downloaded from
vlookup函数的使用方法及实例vlookup函数的使用方法及实例 excel中vlookup函数的应用,重要在于实践。 下面我们先了就下函数的构成;接着举个例子说下;最后总结下急提下遇到的相关问题: (本作者采用的是excel2003版,不过这函数在任何版本都适应) 2首先我们介绍下使用的函数vlookup 的几个参数,vlookup是判断引用数据的函数,它总共有四个参数,依次是: 1、判断的条件 2、跟踪数据的区域 3、返回第几列的数据 4、是否精确匹配 该函数的语法规则如下: =VLOOKUP(lookup_value,table_array,col_index_num,range_looku p) 该函数的语法规则可以查看到,如下图: (excel07版) 如下图,已知表sheet1中的数据如下,如何在数据表二sheet2 中如下引用:当学号随机出现的时候,如何在B列显示其对应的物理成绩? 根据问题的需求,这个公式应该是:
vmdk文件损坏打不开怎么修复vmware vmdk文件损坏打不开修复方法一 EasyRecovery数据恢复软件支持恢复VMDK文件并存储在本地文件系统中。由于数据和有关虚拟服务器的配置信息都存储在VMDK文件中,而每个虚拟系统下通常又有多个VMDK镜像,此时选择正确的VMDK镜像对成功的完成文件恢复扫描而言就显得至关重要了。载入VMDK镜像并选择对应的卷,以开始扫描VMDK文件。 根据EasyRecovery软件给出的提示操作,完成VMDK文件恢复。 当然要想保证VMDK文件恢复的顺利进行,还需注意以下几点: 1、当发现数据丢失之后,不要进行任何操作,因操作系统运行时产生的虚拟内存和临时文件会破坏数据或覆盖数据; 2、不要轻易尝试Windows的系统还原功能,这并不会找回丢失的文件,只会为后期的恢复添置不必要的障碍; 3、不要反复使用杀毒软件,这些操作是无法找回丢失文件的。 vmware vmdk文件损坏打不开修复方法二
硕037班 刘文3110038020 2011/4/20交互式多模型仿真与分析IMM算法与GBP算法的比较,算法实现和运动目标跟踪仿真,IMM算法的特性分析 多源信息融合实验报告
交互式多模型仿真与分析 一、 算法综述 由于混合系统的结构是未知的或者随机突变的,在估计系统状态参数的同时还需要对系统的运动模式进行辨识,所以不可能通过建立起一个固定的模型对系统状态进行效果较好的估计。针对这一问题,多模型的估计方法提出通过一个模型集{}(),1,2,,j M m j r == 中不同模型的切换来匹配不同目标的运动或者同一目标不同阶段的运动,达到运动模式的实时辨识的效果。 目前主要的多模型方法包括一阶广义贝叶斯方法(BGP1),二阶广义贝叶斯方法(GPB2)以及交互式多模型方法等(IMM )。这些多模型方法的共同点是基于马尔科夫链对各自的模型集进行切换或者融合,他们的主要设计流程如下图: M={m1,m2,...mk} K 时刻输入 值的形式 图一 多模型设计方法 其中,滤波器的重初始化方式是区分不同多模型算法的主要标准。由于多模型方法都是基于一个马尔科夫链来切换与模型的,对于元素为r 的模型集{}(),1,2,,j M m j r == ,从0时刻到k 时刻,其可能的模型切换轨迹为 120,12{,,}k i i i k trace k M m m m = ,其中k i k m 表示K-1到K 时刻,模型切换到第k i 个, k i 可取1,2,,r ,即0,k trace M 总共有k r 种可能。再令1 2 1 ,,,,k k i i i i μ+ 为K+1时刻经由轨迹0,k trace M 输入到第1k i +个模型滤波器的加权系数,则输入可以表示为 0,11 2 1 12|,,,,|,,,???k k trace k k k i M k k i i i i k k i i i x x μ++=?∑ 可见轨迹0,k trace M 的复杂度直接影响到算法计算量和存储量。虽然全轨迹的
?哈弗曼编码 A method for the construction of minimum-re-dundancy codes, 耿国华1数据结构1北京:高等教育出版社,2005:182—190 严蔚敏,吴伟民.数据结构(C语言版)[M].北京:清华大学出版社,1997. 冯桂,林其伟,陈东华.信息论与编码技术[M].北京:清华大学出版社,2007. 刘大有,唐海鹰,孙舒杨,等.数据结构[M].北京:高等教育出版社,2001 ?压缩实现 速度要求 为了让它(huffman.cpp)快速运行,同时不使用任何动态库,比如STL或者MFC。它压缩1M数据少于100ms(P3处理器,主频1G)。 压缩过程 压缩代码非常简单,首先用ASCII值初始化511个哈夫曼节点: CHuffmanNode nodes[511]; for(int nCount = 0; nCount < 256; nCount++) nodes[nCount].byAscii = nCount; 其次,计算在输入缓冲区数据中,每个ASCII码出现的频率: for(nCount = 0; nCount < nSrcLen; nCount++) nodes[pSrc[nCount]].nFrequency++; 然后,根据频率进行排序: qsort(nodes, 256, sizeof(CHuffmanNode), frequencyCompare); 哈夫曼树,获取每个ASCII码对应的位序列: int nNodeCount = GetHuffmanTree(nodes); 构造哈夫曼树 构造哈夫曼树非常简单,将所有的节点放到一个队列中,用一个节点替换两个频率最低的节点,新节点的频率就是这两个节点的频率之和。这样,新节点就是两个被替换节点的父
LZ77压缩算法实验报告 一、实验内容 使用C++编程实现LZ77压缩算法的实现。 二、实验目的 用LZ77实现文件的压缩。 三、实验环境 1、软件环境:Visual C++ 6.0 2、编程语言:C++ 四、实验原理 LZ77 算法在某种意义上又可以称为“滑动窗口压缩”,这是由于该算法将一个虚拟的,可以跟随压缩进程滑动的窗口作为术语字典,要压缩的字符串如果在该窗口中出现,则输出其出现位置和长度。使用固定大小窗口进行术语匹配,而不是在所有已经编码的信息中匹配,是因为匹配算法的时间消耗往往很多,必须限制字典的大小才能保证算法的效率;随着压缩的进程滑动字典窗口,使其中总包含最近编码过的信息,是因为对大多数信息而言,要编码的字符串往往在最近的上下文中更容易找到匹配串。 五、LZ77算法的基本流程 1、从当前压缩位置开始,考察未编码的数据,并试图在滑动窗口中找出最长的匹 配字符串,如果找到,则进行步骤2,否则进行步骤3。 2、输出三元符号组( off, len, c )。其中off 为窗口中匹
配字符串相对窗口边 界的偏移,len 为可匹配的长度,c 为下一个字符。然后将窗口向后滑动len + 1 个字符,继续步骤1。 3、输出三元符号组( 0, 0, c )。其中c 为下一个字符。然后将窗口向后滑动 len + 1 个字符,继续步骤1。 六、源程序 /********************************************************************* * * Project description: * Lz77 compression/decompression algorithm. * *********************************************************************/ #include VLOOKUP函数调用方法如下:(本次以提取RRU挂高数据为例) 一、本次涉及的相关文档。 1.《某地区TD宏站现场勘测数据汇总表》如表1-1,共1000多站,本次共列出104个站点的信息: 查看原文档请双击图标:某地区TD宏站现场 查勘数据汇总表,表1-1抓图如下: 2.某工程报价单,共30个宏站,如表1-2(本报价单其他信息均删除,只保留了站点名) 查看原文档请双击图标:某工程报价单.xlsx ,表1-2抓图如下: 二、本次我们以从表1-1中提取表1-2中30个站点的RRU挂高为例,具体步骤如下: 1.先在表1-2中增加“RRU挂高”这一列,然后先提取“某城关水泵厂南”的RRU挂高。操作方法为双击下图所示灰色表格,然后鼠标左键单击列表上面的fx插入函 数。 2.点fx后弹出如下图标,在下拉列表中选择“VLOOKUP”,点确定。 3.点确定后,弹出VLOOKUP函数调用表,包含4个部分(lookup_value、Table_array、C ol_index_num、Range_lookup)。 lookup_value:需要在数据表首列进行搜索的值,本次值为表1-1中的位置B2,用 鼠标单击表1-1中的“某城关水泵厂南”,即可自动输入。。 Table_array:需要在其中搜索数据的信息表,即在表1-2中选择一个搜索区域, 注意所选区域第一列必须是与Lookup_value中查找数值相匹配的 列(本次表1-1中的B列),最后一列必须大于等于RRU挂高那一列 (大于等于C列),至于下拉行数肯定要大于等于106行。如下图: 选择相关区域后,VLOOKUP表中的Table_array会自动输入表1-1中所选区域,如 下图: 实验名称:LZSS压缩算法实验报告 一、实验内容 使用Visual 6..0 C++编程实现LZ77压缩算法。 二、实验目的 用LZSS实现文件的压缩。 三、实验原理 LZSS压缩算法是词典编码无损压缩技术的一种。LZSS压缩算法的字典模型使用了自适应的方式,也就是说,将已经编码过的信息作为字典, 四、实验环境 1、软件环境:Visual C++ 6.0 2、编程语言:C++ 五、实验代码 #include -- need byte-swap function for big endian CPU */ #define READ_LE32(X) *(uint32_t *)(X) #define WRITE_LE32(X,Y) *(uint32_t *)(X) = (Y) /* this assumes sizeof(long)==4 */ typedef unsigned long uint32_t; /* text (input) size counter, code (output) size counter, and counter for reporting progress every 1K bytes */ static unsigned long g_text_size, g_code_size, g_print_count; /* ring buffer of size N, with extra F-1 bytes to facilitate string comparison */ static unsigned char g_ring_buffer[N + F - 1]; /* position and length of longest match; set by insert_node() */ static unsigned g_match_pos, g_match_len; /* left & right children & parent -- these constitute binary search tree */ static unsigned g_left_child[N + 1], g_right_child[N + 257], g_parent[N + 1]; /* input & output files */ static FILE *g_infile, *g_outfile; /***************************************************************************** initialize trees *****************************************************************************/ static void init_tree(void) { unsigned i; /* For i = 0 to N - 1, g_right_child[i] and g_left_child[i] will be the right and left children of node i. These nodes need not be initialized. Also, g_parent[i] is the parent of node i. These are initialized to NIL (= N), which stands for 'not used.' For i = 0 to 255, g_right_child[N + i + 1] is the root of the tree for strings that begin with character i. These are initialized to NIL. Note there are 256 trees. */ for(i = N + 1; i <= N + 256; i++) g_right_child[i] = NIL; for(i = 0; i < N; i++) g_parent[i] = NIL; } /***************************************************************************** Inserts string of length F, g_ring_buffer[r..r+F-1], into one of the trees (g_ring_buffer[r]'th tree) and returns the longest-match position and length via the global variables g_match_pos and g_match_len. If g_match_len = F, then removes the old node in favor of the new one, because the old one will be deleted sooner. excel博大精深,其使用中有许多细节的地方需要注意。 vlookup函数的使用,其语法我就不解释了,百度很多,其实我自己也没看懂语法的解释,下面就按照我自己的理解来说说怎么用的。首先,这个函数是将一个表中的数据导入另一个表中,其中这两个表有一列数据是相同项,但是排列顺序不同。举例说明; 表1 表2 将表1中的face量一列导入表2中,但两表中的名称一列的排列顺序是不同的。此时需要使用vlookup函数。 下面介绍vlookup的使用方法。 将鼠标放到表2中的D2单元格上,点击fx,会出现一个对话框,里面有vlookup函数。若在常用函数里面没有,下拉找“查找与引用”,里面有此函数。点确定。表示此函数是在表2中的D2单元格中应用。 此时出现对话框: 在第个格里输入B2,直接用鼠标在表2中点击B2单元格即可。表示需要在查找的对象是表2中的B2单元格中的内容。 然后是第二个格,点表1,用鼠标选择整个表的所有数据。表示要在表1中的B1—C14区域查找表2中的B2单元格中的内容。 第三个格里输入在表2中要导入的列数在表1中的列数的数字。在此例中为C列,其列数数字为2.表示将表1中(B1—C14)区域中查找到的单元格里的内容相对应的列(第2列)中的单元格中的内容(face量列中的数据)导入表2中相应的单元格(D2)。 最后一个格中输入“0”。表示查找不到就出现#N/A。点确定,即出现相应数据,然后下拉复制格式。 当下拉出现这种情况的时候: 其实是其查找区域在下拉过程中随着行的改变而改变了。需要对查找区域做一下固定。其方法为,在选择区域后,在区域前面加“$”号($B$1:$C$14)。 多媒体数据压缩实验报告 篇一:多媒体实验报告_文件压缩 课程设计报告 实验题目:文件压缩程序 姓名:指导教师:学院:计算机学院专业:计算机科学与技术学号: 提交报告时间:20年月日 四川大学 一,需求分析: 有两种形式的重复存在于计算机数据中,文件压缩程序就是对这两种重复进行了压 缩。 一种是短语形式的重复,即三个字节以上的重复,对于这种重复,压缩程序用两个数字:1.重复位置距当前压缩位置的距离;2.重复的长度,来表示这个重复,假设这两个数字各占一个字节,于是数据便得到了压缩。 第二种重复为单字节的重复,一个字节只有256种可能的取值,所以这种重复是必然的。给 256 种字节取值重新编码,使出现较多的字节使用较短的编码,出现较少的字节使用较长的编码,这样一来,变短的字节相对于变长的字节更多,文件的总长度就会减少,并且,字节使用比例越不均 匀,压缩比例就越大。 编码式压缩必须在短语式压缩之后进行,因为编码式压缩后,原先八位二进制值的字节就被破坏了,这样文件中短语式重复的倾向也会被破坏(除非先进行解码)。另外,短语式压缩后的结果:那些剩下的未被匹配的单、双字节和得到匹配的距离、长度值仍然具有取值分布不均匀性,因此,两种压缩方式的顺序不能变。 本程序设计只做了编码式压缩,采用Huffman编码进行压缩和解压缩。Huffman编码是一种可变长编码方式,是二叉树的一种特殊转化形式。编码的原理是:将使用次数多的代码转换成长度较短的代码,而使用次数少的可以使用较长的编码,并且保持编码的唯一可解性。根据 ascii 码文件中各 ascii 字符出现的频率情况创建 Huffman 树,再将各字符对应的哈夫曼编码写入文件中。同时,亦可根据对应的哈夫曼树,将哈夫曼编码文件解压成字符文件. 一、概要设计: 压缩过程的实现: 压缩过程的流程是清晰而简单的: 1. 创建 Huffman 树 2. 打开需压缩文件 3. 将需压缩文件中的每个 ascii 码对应的 huffman 编码按 bit 单位输出生成压缩文件压缩结束。 VLOOKUP函数的使用方法(入门级) VLOOKUP函数是Excel中几个最重函数之一,为了方便大家学习,兰色幻想特针对VLOOKUP 函数的使用和扩展应用,进行一次全面综合的说明。本文为入门部分 一、入门级 VLOOKUP是一个查找函数,给定一个查找的目标,它就能从指定的查找区域中查找返回想要查找到的值。它的基本语法为: VLOOKUP(查找目标,查找范围,返回值的列数,精确OR模糊查找) 下面以一个实例来介绍一下这四个参数的使用 例1:如下图所示,要求根据表二中的姓名,查找姓名所对应的年龄。 公式:B13 =VLOOKUP(A13,$B$2:$D$8,3,0) 参数说明: 1 查找目标:就是你指定的查找的内容或单元格引用。本例中表二A列的姓名就是查找目标。我们要根据表二的“姓名”在表一中A列进行查找。 公式:B13 =VLOOKUP(A13,$B$2:$D$8,3,0) 2 查找范围(VLOOKUP(A13,$B$2:$D$8,3,0) ):指定了查找目标,如果没有说从哪里查找,EXCEL肯定会很为难。所以下一步我们就要指定从哪个范围中进行查找。VLOOKUP的这第二个参数可以从一个单元格区域中查找,也可以从一个常量数组或内存数组中查找。本例中要从表一中进行查找,那么范围我们要怎么指定呢?这里也是极易出错的地方。大家一定要注意,给定的第二个参数查找范围要符合以下条件才不会出错: A 查找目标一定要在该区域的第一列。本例中查找表二的姓名,那么姓名所对应的表一的姓名列,那么表一的姓名列(列)一定要是查找区域的第一列。象本例中,给定的区域要从第二列开始,即$B$2:$D$8,而不能是$A$2:$D$8。因为查找的“姓名”不在$A$2:$D$8区域的第一列。 B 该区域中一定要包含要返回值所在的列,本例中要返回的值是年龄。年龄列(表一的D列)一定要包括在这个范围内,即:$B$2:$D$8,如果写成$B$2:$C$8就是错的。 3 返回值的列数(B13 =VLOOKUP(A13,$B$2:$D$8,3,0))。这是VLOOKUP第3个参数。它是一个整数值。它怎么得来的呢。它是“返回值”在第二个参数给定的区域中的列数。本例中我们 价值工程 置,是一项十分有意义的工作。另外恶意代码的检测和分析是一个长期的过程,应对其新的特征和发展趋势作进一步研究,建立完善的分析库。 参考文献: [1]CNCERT/CC.https://www.doczj.com/doc/0a4367231.html,/publish/main/46/index.html. [2]LO R,LEVITTK,OL SSONN R.MFC:a malicious code filter [J].Computer and Security,1995,14(6):541-566. [3]KA SP ER SKY L.The evolution of technologies used to detect malicious code [M].Moscow:Kaspersky Lap,2007. [4]LC Briand,J Feng,Y Labiche.Experimenting with Genetic Algorithms and Coupling Measures to devise optimal integration test orders.Software Engineering with Computational Intelligence,Kluwer,2003. [5]Steven A.Hofmeyr,Stephanie Forrest,Anil Somayaji.Intrusion Detection using Sequences of System calls.Journal of Computer Security Vol,Jun.1998. [6]李华,刘智,覃征,张小松.基于行为分析和特征码的恶意代码检测技术[J].计算机应用研究,2011,28(3):1127-1129. [7]刘威,刘鑫,杜振华.2010年我国恶意代码新特点的研究.第26次全国计算机安全学术交流会论文集,2011,(09). [8]IDIKA N,MATHUR A P.A Survey of Malware Detection Techniques [R].Tehnical Report,Department of Computer Science,Purdue University,2007. 0引言 现有的压缩算法有很多种,但是都存在一定的局限性,比如:LZw [1]。主要是针对数据量较大的图像之类的进行压缩,不适合对简单报文的压缩。比如说,传输中有长度限制的数据,而实际传输的数据大于限制传输的数据长度,总体数据长度在100字节左右,此时使用一些流行算法反而达不到压缩的目的,甚至增大数据的长度。本文假设该批数据为纯数字数据,实现压缩并解压缩算法。 1数据压缩概念 数据压缩是指在不丢失信息的前提下,缩减数据量以减少存储空间,提高其传输、存储和处理效率的一种技术方法。或按照一定的算法对数据进行重新组织,减少数据的冗余和存储的空间。常用的压缩方式[2,3]有统计编码、预测编码、变换编码和混合编码等。统计编码包含哈夫曼编码、算术编码、游程编码、字典编码等。 2常见几种压缩算法的比较2.1霍夫曼编码压缩[4]:也是一种常用的压缩方法。其基本原理是频繁使用的数据用较短的代码代替,很少使用 的数据用较长的代码代替,每个数据的代码各不相同。这些代码都是二进制码,且码的长度是可变的。 2.2LZW 压缩方法[5,6]:LZW 压缩技术比其它大多数压缩技术都复杂,压缩效率也较高。其基本原理是把每一个第一次出现的字符串用一个数值来编码,在还原程序中再将这个数值还成原来的字符串,如用数值0x100代替字符串ccddeee"这样每当出现该字符串时,都用0x100代替,起到了压缩的作用。 3简单报文数据压缩算法及实现 3.1算法的基本思想数字0-9在内存中占用的位最 大为4bit , 而一个字节有8个bit ,显然一个字节至少可以保存两个数字,而一个字符型的数字在内存中是占用一个字节的,那么就可以实现2:1的压缩,压缩算法有几种,比如,一个自己的高四位保存一个数字,低四位保存另外一个数字,或者,一组数字字符可以转换为一个n 字节的数值。N 为C 语言某种数值类型的所占的字节长度,本文讨论后一种算法的实现。 3.2算法步骤 ①确定一种C 语言的数值类型。 —————————————————————— —作者简介:安建梅(1981-),女,山西忻州人,助理实验室,研究方 向为软件开发与软交换技术;季松华(1978-),男,江苏 南通人,高级软件工程师,研究方向为软件开发。 数据快速压缩算法的研究以及C 语言实现 The Study of Data Compression and Encryption Algorithm and Realization with C Language 安建梅①AN Jian-mei ;季松华②JI Song-hua (①重庆文理学院软件工程学院,永川402160;②中信网络科技股份有限公司,重庆400000)(①The Software Engineering Institute of Chongqing University of Arts and Sciences ,Chongqing 402160,China ; ②CITIC Application Service Provider Co.,Ltd.,Chongqing 400000,China ) 摘要:压缩算法有很多种,但是对需要压缩到一定长度的简单的报文进行处理时,现有的算法不仅达不到目的,并且变得复杂, 本文针对目前一些企业的需要,实现了对简单报文的压缩加密,此算法不仅可以快速对几十上百位的数据进行压缩,而且通过不断 的优化,解决了由于各种情况引发的解密错误,在解密的过程中不会出现任何差错。 Abstract:Although,there are many kinds of compression algorithm,the need for encryption and compression of a length of a simple message processing,the existing algorithm is not only counterproductive,but also complicated.To some enterprises need,this paper realizes the simple message of compression and encryption.This algorithm can not only fast for tens of hundreds of data compression,but also,solve the various conditions triggered by decryption errors through continuous optimization;therefore,the decryption process does not appear in any error. 关键词:压缩;解压缩;数字字符;简单报文Key words:compression ;decompression ;encryption ;message 中图分类号:TP39文献标识码:A 文章编号:1006-4311(2012)35-0192-02 ·192· VLOOKUP函数是Excel中的一个纵向查找函数,它与LOOKUP函数和HLOOKUP函数属于一类函数,在工作中都有广泛应用。VLOOKUP是按列查找,最终返回该列所需查询列序所对应的值;与之对应的HLOOKUP是按行查找的。 VLOOKUP函数的语法结构 整个计算机就相当于一门语言,首先我们就是要获取该函数的语法结构。以下是官网的语法结构 VLOOKUP(lookup_value, table_array, col_index_num, [range_looku p])。 书上表述就是VLOOKUP(查找值,查找范围,查找列数,精确匹配或者近似匹配) 在我们的工作中,几乎都使用精确匹配,该项的参数一定要选择为false。否则返回值会出乎你的意料。 VLOOKUP函数使用示范 vlookup就是竖直查找,即列查找。通俗的讲,根据查找值参数,在查找范围的第一列搜索查找值,找到该值后,则返回值为:以第一列为准,往后推数查找列数值的这一列所对应的值。这也是为什么该函数叫做vlookup(v为vertic al-竖直之意,lookup即时英文的查找之意)。 现有如下手机的每日销售毛数据(图左),A分销商需要提供四个型号的销售数据(图右) 这个时候,你大概可能回去一个一个人工查找,因为我所提供的数据数量很少,但是其实工作中这种数据很庞大的,人工查找无疑即浪费时间,而且不能让A分销商相信你所提供数据的准确性。接下来,我们就需要本次的主角登场了。使用vlookup函数。 第一步:选中要输入数据的单元格,=VLOOKUP(H3,$A$3:$F$19,5,FALSE)如图 压缩编码算法设计与实现实验报告 一、实验目的:用C语言或C++编写一个实现Huffman编码的程序,理解对数据进行无损压缩的原理。 二、实验内容:设计一个有n个叶节点的huffman树,从终端读入n个叶节 点的权值。设计出huffman编码的压缩算法,完成对n个节点的编码,并写出程序予以实现。 三、实验步骤及原理: 1、原理:Huffman算法的描述 (1)初始化,根据符号权重的大小按由大到小顺序对符号进行排序。 (2)把权重最小的两个符号组成一个节点, (3)重复步骤2,得到节点P2,P3,P4……,形成一棵树。 (4)从根节点开始顺着树枝到每个叶子分别写出每个符号的代码 2、实验步骤: 根据算法原理,设计程序流程,完成代码设计。 首先,用户先输入一个数n,以实现n个叶节点的Huffman 树; 之后,输入n个权值w[1]~w[n],注意是unsigned int型数值; 然后,建立Huffman 树; 最后,完成Huffman编码。 四、实验代码:#include int min(HuffmanTree t,int i) { int j,flag; unsigned int k = MAX; for(j=1;j<=i;j++) if(t[j].parent==0&&t[j].weight VLOOKUP函数的使用方法(入门级)一、入门级 VLOOKUP是一个查找函数,给定一个查找的目标,它就能从指定的查找区域中查找返回想要查找到的值。它的基本语法为: VLOOKUP(查找目标,查找范围,返回值的列数,精确OR模糊查找) 下面以一个实例来介绍一下这四个参数的使用 例1:如下图所示,要求根据表二中的姓名,查找姓名所对应的年龄。 公式:B13 =VLOOKUP(A13,$B$2:$D$8,3,0) 参数说明: 1 查找目标:就是你指定的查找的内容或单元格引用。本例中表二A列的姓名就是查找目标。我们要根据表二的“姓名”在表一中A列进行查找。 公式:B13 =VLOOKUP(A13,$B$2:$D$8,3,0) 2 查找范围(VLOOKUP(A13,$B$2:$D$8,3,0) ):指定了查找目标,如果没有说从哪里查找,EXCEL肯定会很为难。所以下一步我们就要指定从哪 个范围中进行查找。VLOOKUP的这第二个参数可以从一个单元格区域中查找, 也可以从一个常量数组或内存数组中查找。本例中要从表一中进行查找,那么范 围我们要怎么指定呢?这里也是极易出错的地方。大家一定要注意,给定的第二 个参数查找范围要符合以下条件才不会出错: A 查找目标一定要在该区域的第一列。本例中查找表二的姓名,那么姓名 所对应的表一的姓名列,那么表一的姓名列(列)一定要是查找区域的第一列。 象本例中,给定的区域要从第二列开始,即$B$2:$D$8,而不能是$A$2:$D$8。 因为查找的“姓名”不在$A$2:$D$8区域的第一列。 B 该区域中一定要包含要返回值所在的列,本例中要返回的值是年龄。年 龄列(表一的D列)一定要包括在这个范围内,即:$B$2:$D$8,如果写成$B $2:$C$8就是错的。 3 返回值的列数(B13 =VLOOKUP(A13,$B$2:$D$8,3,0))。这是VLO OKUP第3个参数。它是一个整数值。它怎么得来的呢。它是“返回值”在第 二个参数给定的区域中的列数。本例中我们要返回的是“年龄”,它是第二个参 数查找范围$B$2:$D$8的第3列。这里一定要注意,列数不是在工作表中的列 数(不是第4列),而是在查找范围区域的第几列。如果本例中要是查找姓名所 交互式多模型算法卡尔曼滤波仿真 1 模型建立 分别以加速度u=0、1、2代表三个不同的运动模型 状态方程x(k+1)=a*x(k)+b*w(k)+d*u 观察方程z(k)=c*x(k)+v(k) 其中,a=[1 dt;0 1],b=[dt^2/2;dt],d=[dt^2/2;dt],c=[1 0] 2 程序代码 由两个功能函数组成,imm_main用来实现imm 算法,move_model1用来生成仿真数据,初始化运动参数 function imm_main %交互式多模型算法主程序 %初始化有关参数 move_model %调用运动模型初始化及仿真运动状态生成函数 load movedata %调入有关参数初始值(a b d c u position velocity pmeas dt tg q r x_hat p_var) p_tran=[0.8 0.1 0.1;0.2 0.7 0.1;0.1 0.2 0.7];%转移概率 p_pri=[0.1;0.6;0.3];%模型先验概率 n=1:2:5; %提取各模型方差矩阵 k=0; %记录仿真步数 like=[0;0;0];%视然函数 x_hat_hat=zeros(2,3);%三模型运动状态重初始化矩阵 u_=zeros(3,3);%混合概率矩阵 c_=[0;0;0];%三模型概率更新系数 %数据保存有关参数初始化 phat=[];%保存位置估值 vhat=[];%保存速度估值 xhat=[0;0];%融合和运动状态 z=0;%量测偏差(一维位置) pvar=zeros(2,2);%融合后估计方差 for t=0:dt:tg; %dt为为仿真步长;tg为仿真时间长度 k=k+1;%记录仿真步数 ct=0; %三模型概率更新系数 c_max=[0 0 0];%混合概率规范系数 p_var_hat=zeros(2,6);%方差估计重初始化矩阵, %[x_hat_hat p_var_hat]=model_reinitialization(p_tran,p_pri,x_hat,p_var);%调用重初始化函数,进行混合估计,生成三个模型重初始化后的运动状态、方差 %混合概率计算 for i=1:3 u_(i,:)=p_tran(i,:)*p_pri(i); end for i=1:3 c_max=c_max+u_(i,:); end 目 录 实验一用C/C++语言实现游程编码 实验二用C/C++语言实现算术编码 实验三用C/C++语言实现LZW编码 实验四用C/C++语言实现2D-DCT变换13 实验一用C/C++语言实现游程编码 1. 实验目的 1) 通过实验进一步掌握游程编码的原理; 2) 用C/C++语言实现游程编码。 2. 实验要求 给出数字字符,能正确输出编码。 3. 实验内容 现实中有许多这样的图像,在一幅图像中具有许多颜色相同的图块。在这些图块中,许多行上都具有相同的颜色,或者在一行上有许多连续的象素都具有相同的颜色值。在这种情况下就不需要存储每一个象素的颜色值,而仅仅存储一个象素的颜色值,以及具有相同颜色的象素数目就可以,或者存储一个象素的颜色值,以及具有相同颜色值的行数。这种压缩编码称为游程编码,常用(run length encoding,RLE)表示,具有相同颜色并且是连续的象素数目称为游程长度。 为了叙述方便,假定一幅灰度图像,第n行的象素值为: 用RLE编码方法得到的代码为:0@81@38@501@40@8。代码中用黑体表示的数字是游程长度,黑体字后面的数字代表象素的颜色值。例如黑体字50代表有连续50个象素具有相同的颜色值,它的颜色值是8。 对比RLE编码前后的代码数可以发现,在编码前要用73个代码表示这一行的数据,而编码后只要用11个代码表示代表原来的73个代码,压缩前后的数据量之比约为7:1,即压缩比为7:1。这说明RLE确实是一种压缩技术,而且这种编码技术相当直观,也非常经济。RLE所能获得的压缩比有多大,这主要是取决于图像本身的特点。如果图像中具有相同颜色的图像块越大,图像块数目越少,获得的压缩比就越高。反之,压缩比就越小。 译码时按照与编码时采用的相同规则进行,还原后得到的数据与压缩前的数据完全相同。因此,RLE是无损压缩技术。 VLOOKUP函数 使用举例 如图 vlookup函数示例 所示,我们要在A2:F12区域中提取100003、100004、100005、100007、100010五人的全年总计销量,并对应的输入到I4:I8中。一个一个的手动查找在数据量大的时候十分繁琐,因此这里使用VLOOKUP函数演示: 首先在I4单元格输入“=Vlookup(”,此时Excel就会提示4个参数。 Vlookup结果演示 第一个参数,很显然,我们要让100003对应的是I4,这里就输入“H4,” ; 第二个参数,这里输入我们要查找的区域(绝对引用),即“$A$2:$F$12,”; 第三个参数,“全年总计”是区域的第六列,所以这里输入“6”,输入“5”就会输入第四季度的项目了; 第四个参数,因为我们要精确的查找工号,所以留空即可。 最后补全最后的右括号“)”,得到公式“=VLOOKUP(H4,$A$2:$F$12,6)”,使用填充柄填充其他单元格即可完成查找操作。 VLOOKUP函数使用注意事项 说到VLOOKUP函数,相信大家都会使用,而且都使用得很熟练了。不过,有几个细节问题,大家在使用时还是留心一下的好。 一.VLOOKUP的语法 VLOOKUP函数的完整语法是这样的: VLOOKUP(lookup_value,table_array,col_index_num,range_lookup) 1.括号里有四个参数,是必需的。最后一个参数range_lookup是个逻辑值,我们常常输入一个0字,或者False;其实也可以输入一个1字,或者true。两者有什么区别呢?前者表示的是完整寻找,找不到就传回错误值#N/A;后者先是找一模一样的,找不到再去找很接近的值,还找不到也只好传回错误值#N/A。这对我们其实也没有什么实际意义,只是满足好奇而已,有兴趣的朋友可以去体验体验。 2.Lookup_value是一个很重要的参数,它可以是数值、文字字符串、或参照地址。我们常常用的是参照地址。用这个参数时,有三点要特别提醒:A)参照地址的单元格格式类别与去搜寻的单元格格式的类别要一致,否则的话有时明明看到有资料,就是抓不过来。特别是参照地址的值是数字时,最为明显,若搜寻的单元格格式类别为文字,虽然看起来都是123,但是就是抓不出东西来的。VLOOKUP函数的使用方法(图解说明_很详细)
LZSS压缩算法实验报告
excel中的vlookup函数的使用方法及注意事项
多媒体数据压缩实验报告
VLOOKUP函数的使用方法(从入门到精通)
数据快速压缩算法的C语言实现
vlookup函数的使用方法实例
压缩编码算法设计与实现实验报告
VLOOKUP函数地使用方法
交互式多模型算法卡尔曼滤波仿真
数据压缩实验指导书
vlookup函数使用说明
相关主题
文本预览